the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Nov 2020
| 11 Nov 2020
Permafrost thawing exhibits a greater influence on bacterial richness and community structure than permafrost age in Arctic permafrost soils
Chao Liang
Tianqi Zhou
Hongzeng Jia
Xiaobin Dong
Global warming accelerates permafrost thawing and changes its microbial community structure, but little is known about how microorganisms in permafrost with different ages respond to thawing. Herein, we disentangled the relative importance of permafrost age (young, medium-aged, old, and ancient, spanning from 50 to 5000 years) and thawing status (active, transitional, and permanently frozen) in shaping bacterial community structure using HiSeq sequencing of the 16S rRNA gene. Our results revealed significant influences of both permafrost thawing and age on bacterial richness. The bacterial richness was significantly higher in the young and thawed permafrost, and the richness increase was mainly observed in Firmicutes, Actinobacteria, Chloroflexi, Deltaproteobacteria, and Alphaproteobacteria. Permafrost thawing led to a gradual change in bacterial community structure and increased contribution of determinism. Permutational analysis of variance demonstrated that thawing significantly changed bacterial community structure at all soil ages, but the community convergence due to permafrost thawing was not observed. Structural equation modeling revealed that permafrost thawing exhibited a greater influence on both bacterial richness and community structure than permafrost age. Our results indicate that microorganisms in permafrost with different ages respond differently to thawing, which eventually leads to distinct bacterial community compositions and different organic carbon decomposition processes in Arctic permafrost.
- Article
(1628 KB) - Full-text XML
-
Supplement
(625 KB) - BibTeX
- EndNote





Global warming accelerates permafrost thawing, and 200 billion tons of carbon is estimated to be released into the atmosphere from global permafrost over the next 300 years (Turetsky et al., 2019). The degradation of soil organic carbon (SOC) is predominately driven by microorganisms (Frank–Fahle et al., 2014), and the quality and quantity of SOC also control the abundance and community structure of the microbial community (Chen et al., 2016). It has been reported that permafrost of different thawing status and ages exhibits distinct labile and recalcitrant carbon quantities, with higher carbohydrates in relation to aliphatic carbon in older than in younger permafrost (Chen et al., 2016; Mueller et al., 2015; Yang et al., 2009). Thus, the distinct SOC composition may subsequently impact the microbial community structure in permafrost soil, and the distinct microbial community structure may respond differently to permafrost thawing. However, the impacts of permafrost age and its interaction with thawing on the microbial community remain largely elusive.
In addition to permafrost age, the contrasting composition of labile and recalcitrant carbons was also reported in the thawed and frozen permafrost. This was proposed to be due to the distinct microbial transformation process in the different permafrost thawing status (Mueller et al., 2015). The microbes in the frozen permafrost are predominately in a state of reduced metabolism rate (Gilichinskii, 1995); thus labile carbon is protected from microbial degradation (Hobbie et al., 2000). In contrast, permafrost thawing substantially activates a diverse range of oligotrophic and copiotrophic bacteria and enriches carbohydrate transporter and metabolism-related genes (Schostag et al., 2019). This leads to an increased bacterial richness and converged community metabolic functions, and the soil carbon being dominated by aliphatic carbon resulted from microbial transformation (Deng et al., 2015; Mackelprang et al., 2011; Monteux et al., 2018; Schostag et al., 2019).
Soil development leads to changes in bacterial community structure, predominately due to nutrient accumulation and vegetation colonization (Bardgett and Walker, 2004; Park et al., 2011). Distinct bacterial community structure has been reported in soils of different ages. For example, young soils in the deglaciation chronosequence exhibit significantly lower bacterial richness than aged soils, and autotrophs play a major role in the accumulation of nutrients (Kazemi et al., 2016; Kim et al., 2017; Liu et al., 2016). In contrast, aged soils with vegetation are dominated by heterotrophs, such as Acidobacteria and Actinobacteria (Kwon et al., 2015). However, little is known about the influence of permafrost age on soil microbial community.
To explore the effects of permafrost age on the response of bacteria to permafrost thawing, the soil bacterial community in Arctic permafrost was characterized using the Illumina sequencing targeting the 16S rRNA gene. Given the continuously changed bacterial community with increasing soil age (Kazemi et al., 2016; Kim et al., 2017), we hypothesized that bacterial richness and community structure would also significantly differ in the permafrost of various ages and respond differently to permafrost thawing. Approximately 20 % of the Arctic coastal plains of northern Alaska contain thaw lakes drained at various stages since the mid-Holocene, which were then developed into ice-rich permafrost (Hinkel et al., 2003). These drained lake basins contain soils ranging from freshly developed organic layers on sediments to fully developed ancient permafrost soils (Mueller et al., 2015). By using the drained thaw lake basin age as a proxy for the time of permafrost formation, it provides an opportunity to investigate the influence of permafrost age on microbial community during permafrost degradation. An earlier study at this site has revealed a high abundance of Candidatus Methanoflorens archaeon in the community (Kao-Kniffin et al., 2015), but how the bacteria in the permafrost of various ages would respond to thawing remains less understood. Thus, we investigated the interactive influence of permafrost thawing and age on permafrost soil bacterial community.
2.1 Site description
The permafrost was sampled in the Barrow Peninsula between 71∘20′ and 71∘27′ N latitude and between 156∘4′ and 156∘7′ W longitude (Kao-Kniffin et al., 2015). Barrow Peninsula is located at the northernmost coast of Alaska and is part of the Arctic Coastal Plain with continuous permafrost. The mean annual temperature is −12 ∘C, and the mean annual precipitation is 104 mm (Mueller et al., 2015). In brief, 16 soil cores were collected along a chronosequence of drained lake basins, spanning in age from young (<50 years old), medium-aged (<300 years old), old (<3000 years old), to ancient (3000–5000 years old) in April 2010. The chronosequence was determined by the degree of plant community succession and 14C carbon dating (Hinkel et al., 2003). At each lake basin, a soil core was collected using a SIPRE corer measuring 80 to 150 cm long and 7.5 cm diameter attached to a Big Beaver earth drill apparatus (Litter Beaver, Inc., Livingstone, TX, USA) mounted on a sled. Each soil core contained three layers: active, transitional, and permanently frozen. The active layer represents the surface soil layer that thaws and refreezes on an annual basis; the transitional layer remains frozen but occasionally thaws during warmer summers; the permanently frozen layer remains annually frozen (Kao-Kniffin et al., 2015). The surface organic layer thickness varies with permafrost age, which is <5, 10–15, 15–30, and 40–50 cm for the young, medium-aged, old, and ancient permafrost (Kao-Kniffin et al., 2015). For each permafrost age–layer combination, there were four sampling replicates. The frozen soil cores were cut with a chop saw into sections of soils horizons in a cold room in Barrow, and soils were homogenized, stored, and transported at −20 ∘C until processed (Mueller et al., 2015). Due to sample quantity limitation, two of the most important soil physicochemical properties, total organic carbon (TOC) and total nitrogen (TN), were measured using dry combustion (Vario MAX CNS Analyzer, Elementar, Hanau, Germany). These factors have been reported to substantially vary in samples with different permafrost ages and thawing statuses (Mueller et al., 2015). For other soil properties and soil profile descriptions please see Kao-Kniffin et al. (2015).
2.2 DNA extraction and sequencing
Total DNA was extracted using the MO BIO Power Soil DNA extraction kit (Mo Bio Laboratories, Carlsbad, CA, USA) according to the manufacturer's instructions. A universal primer set to 515F (5'-GTGCCAGCMGCCGCGGTAA-3') and 806r (5'-GGACTACHVGGGTWTCTAAT-3') with 12-nucleotide-long unique barcodes was used to amplify the V4 hyper-variable region of the 16S rRNA gene (Caporaso et al., 2012). The PCR mixture (25 µL) contained 1x PCR, 1.5 mM of MgCl2, 0.4 µM of each deoxynucleoside triphosphate base, 1.0 µM of each primer, 0.5 U of Ex Taq (TaKaRa, Dalian, China), and 20 ng of a DNA template. The PCR amplification program included an initial denaturation at 94 ∘C for 3 min, followed by 30 cycles of 94 ∘C for 20 s, 56 ∘C for 30 s, 72 ∘C for 45 s, and a final extension at 72 ∘C for 10 min. PCR products were pooled in equal molar amounts and then used for paired-end sequencing (2×250 base pairs) on the Illumina HiSeq 2500 sequencer at the Magigene Biotechnology Co Ltd (Guangzhou, China).
2.3 Data processing
Three samples generated very low reads, and to avoid artifacts from different sequencing batches, these three samples were removed from the downstream analysis. Raw sequence data were processed using the MOTHUR v. 1.34.3 (Schloss et al., 2009). Paired-end reads were merged and quality screened with the following settings: as the amplicon size was approximately 300 base pairs, sequences with length <250 or >350, more than 1 mismatch in the primer region, average quality <30, ambiguous bases >0, and homopolymer length >9 were removed from the subsequent downstream analysis. The sequences were then aligned against the Silva reference alignment (release 128), which was trimmed to include only the same region amplified, and those sequences that did not align were removed. Chimeric sequences were identified using the UCHIME (Edgar et al., 2011) and removed. The remaining sequences were classified using the Bayesian classifier against the Silva database (release 128), with a minimum confidence score of 80 % (Wang et al., 2007), and all Eukaryota, chloroplasts, mitochondria, and unknown sequences were removed. Archaeal sequences were also removed to concentrate the study on the bacterial community. Finally, sequences were classified into operational taxonomic units (OTUs) at 97 % identity, and singletons were then removed. The dataset was sub-sampled to an equal size of 16 144, which was the smallest sample size across the entire dataset. Bacterial richness, evenness, and Shannon diversity indices were calculated using the summary.single command in the mothur program (Schloss et al., 2009). A bacterial phylogenetic diversity index was calculated using the pd command in the Picante package (Kembel et al., 2010) under the R environment.
2.4 Statistical analysis
Bacterial richness, evenness, Shannon diversity, phylogenetic diversity, total organic carbon, and total nitrogen in the samples with different permafrost ages and thawing statuses were compared using the two-way ANOVA, and pairwise differences were assessed using the Tukey's HSD test. The Levene's test was used to ensure the homogeneity of variances for the dependent variables (bacterial richness, TOC, and TN) for each combination of the independent variables (Brown and Forsythe, 1974). One-way ANOVA was used to examine the significance of the differences among the permafrost soils of different thawing statuses within the same age class. Both two-way ANOVA and one-way ANOVA were performed using SPSS 23 (SPSS Inc., Armonk, NY, USA).
A non-metric multidimensional scaling (NMDS) plot was generated from the Hellinger-transformed bacterial community dataset based on the Bray–Curtis dissimilarity matrix using Primer 6 (Clarke and Warwick, 2006). The contributions of carbon, nitrogen, C:N ratio, permafrost age, and thawing status to the community structure were quantified using the distance-based linear model (DistLM) after normalization. Permutational analysis of variance (PERMANOVA) was used to examine the influence of permafrost thawing status and age on bacterial community structure (Anderson, 2001) using Primer 6. We compared the multivariate dispersion homogeneity to assess the bacterial community convergence by permafrost thawing status, using permutational analysis of multivariate dispersions (PERMDISP) (Anderson, 2006).
2.5 Quantifying the contribution of stochasticity
A Bray–Curtis dissimilarity-based normalized stochasticity ratio (NST) index was calculated using the “NST” package in R (https://www.r-project.org/, last access: 30 September 2019) to represent the contribution of stochasticity to community assembly (Ning et al., 2019). The NST index values range from 0 % to 100 %: 0 % indicates zero contribution of stochasticity, whereas 100 % indicates the community assembly being completely stochasticity-driven.
2.6 Structural equation modeling (SEM) analysis
SEM was conducted using AMOS 21 software (IBM SPSS Inc., Chicago, IL, USA) to assess the relative importance of permafrost thawing status and age in shaping bacterial richness and community structure. The permafrost age was ranked from 1 to 4 for the youngest to the oldest permafrost soils, whereas the thawing status was ranked from 1 to 3 for the active to frozen permafrost. The standardized regression weights were calculated for the bacterial richness and the first and second axis coordinates of the NMDS ordination plot. The goodness of fit for the model was judged by the following measures (Guo et al., 2015): (1) comparative fit index (CFI), in which the model has a good fit when 0.97 ≤ CFI ≤ 1.00 and an acceptable fit when 0.95 ≤ CFI < 0.97; (2) goodness-of-fit index (GFI), in which the model has a good fit when 0.95 ≤ GFI ≤ 1.00 and acceptable fit when 0.90 ≤ GFI < 0.95; (3) normed fit index (NFI), in which the model has a good fit when 0.95 ≤ NFI ≤ 1.00 and an acceptable fit when 0.90≤ NFI < 0.95; and (4) χ2 test, in which the model has a good fit when 0≤χ2 with degrees of freedom ≤2 and 0.05 <P≤ 1.00, and an acceptable fit when 2<χ2 with degrees of freedom ≤3 and ; and the root mean square error of approximation (RMSEA, the model has a good fit when 0 ≤ RMSEA ≤ 0.05 and , and an acceptable fit when 0.05 < RMSEA ≤ 0.08 and 0.05 0.10). The relative contributions of TOC, TN, permafrost age, and thawing status to the richness and community structure were also evaluated by the random forest approach using the rfPermute package for R (Archer, 2016).
3.1 The influence of permafrost age and thawing status on soil organic carbon and nitrogen
Across all samples, soil TOC ranged from 0.5 % to 35.6 % and exhibited significant differences by permafrost thawing status (two-way ANOVA, P<0.01; Fig. 1a), but not by permafrost age (P=0.343; Fig. 1b). The active layer soil exhibited the highest TOC (16.7 %) and was significantly higher than the permanently frozen layer soil (5.6 %, Tukey's HSD P<0.001). Soil TN ranged from 0.1 % to 1.5 %, and significant differences were only detected by permafrost thawing status (P=0.007; Fig. 1c), but not by permafrost age (P=0.446; Fig. 1d). The active layer soil exhibited the highest TN (0.73 %) and was significantly higher than the permanently frozen layer soil (0.29 %, Tukey's HSD, P=0.004).
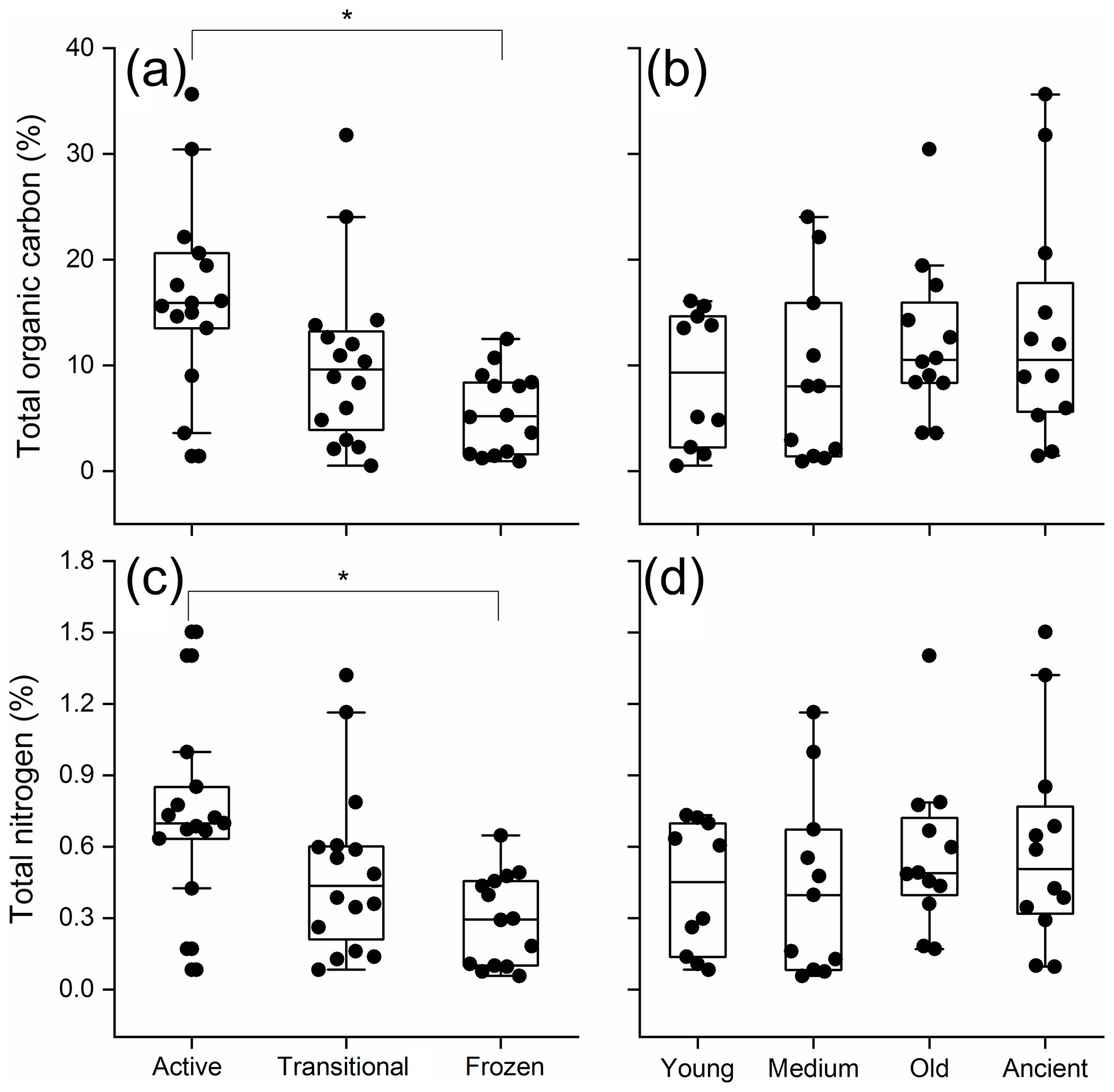
Figure 1Total organic carbon (a and b) and total nitrogen (c and d) with the permafrost age (young, medium-aged, old, and ancient) and permafrost thawing status (active, transitional, and permanently frozen).
3.2 The influence of permafrost age and thawing status on bacteria richness
A total of 1 679 607 bacterial sequences were retained, with an average sequence length of 292 base pairs. After rarefying to an equal sequencing depth, there were 2415 bacterial OTUs at the 97 % nucleic acid sequence identity retained, and the community was dominated by Firmicutes (42 %), Actinobacteria (28.9 %), and Proteobacteria (10.6 %; Fig. S1).
Our results exhibited substantial differences in the bacterial richness among the permafrost soils of different thawing status (two-way ANOVA, P<0.001; Fig. 2a, Table S1) and ages (P=0.013; Fig. 2b). A significantly higher bacterial richness was observed in the active layer soil (358 OTUs) than the transitional (287 OTUs; Pairwise Tukey's HSD tests, P=0.011) and the frozen layer soils (248 OTUs, P<0.001, Table S2). Young permafrost (380 OTUs) exhibited a significantly higher bacterial richness than the medium-aged (265 OTUs, P=0.001), old (287 287, P=0.002), and ancient soils (271 OTUs, P=0.009, Table S3). In comparison, the influence of permafrost age and thawing status on bacteria Shannon diversity was non-significant (two-way ANOVA, P=0.058 and 0.53, respectively; Fig. S2). This differed from the phylogenetic diversity, where significant influence was observed for age (P=0.015) and thawing (P=0.001).

Figure 2Bacterial richness with the permafrost thawing status (a) and age (b). The richness is indicated by operational taxonomic unit (OTU) number. Different letters indicate significant difference at P<0.05. Young, medium-aged, old, and ancient are permafrost soil ages; active, transitional, and permanently frozen are permafrost thawing status.
The influence of permafrost thawing on bacterial richness was only significantly observed in the young permafrost (one-way ANOVA, P<0.001; Fig. 2b, Table S4), whereas those in the medium-aged, old, and ancient soils were non-significant (P=0.445, 0.48, and 0.35, respectively). In the young permafrost, permafrost thawing significantly increased OTU number from 248 in the frozen layer soil to 471 in the active layer soil (Table S5). The increased bacterial richness was mainly attributed to the significant increase detected in Firmicutes (ANOVA, P=0.011), Actinobacteria (P=0.002), Chloroflexi (P=0.002), Deltaproteobacteria (P=0.02), and Alphaproteobacteria (P=0.008; Table S6).
3.3 The influence of permafrost thawing status and age on bacterial community structure
A Bray–Curtis distance-based NMDS ordination plot revealed a clear separation of the bacterial community structure by permafrost thawing status (Fig. 3a), while the separation by permafrost age was less obvious (Fig. 3b). The results of DistLM analyses revealed that the measured soil factors, thawing status, and age explained a total of 10.7 % of the bacterial community structure. TN was the most important factor, explaining 7.2 % of the community structure (P=0.001). This was followed by C:N ratio, TOC, soil age, and thawing status, which additionally explained 3.5 % (P=0.028), 3 % (P=0.083), 2.9 % (P=0.105), and 2.8 % (P=0.111), respectively.
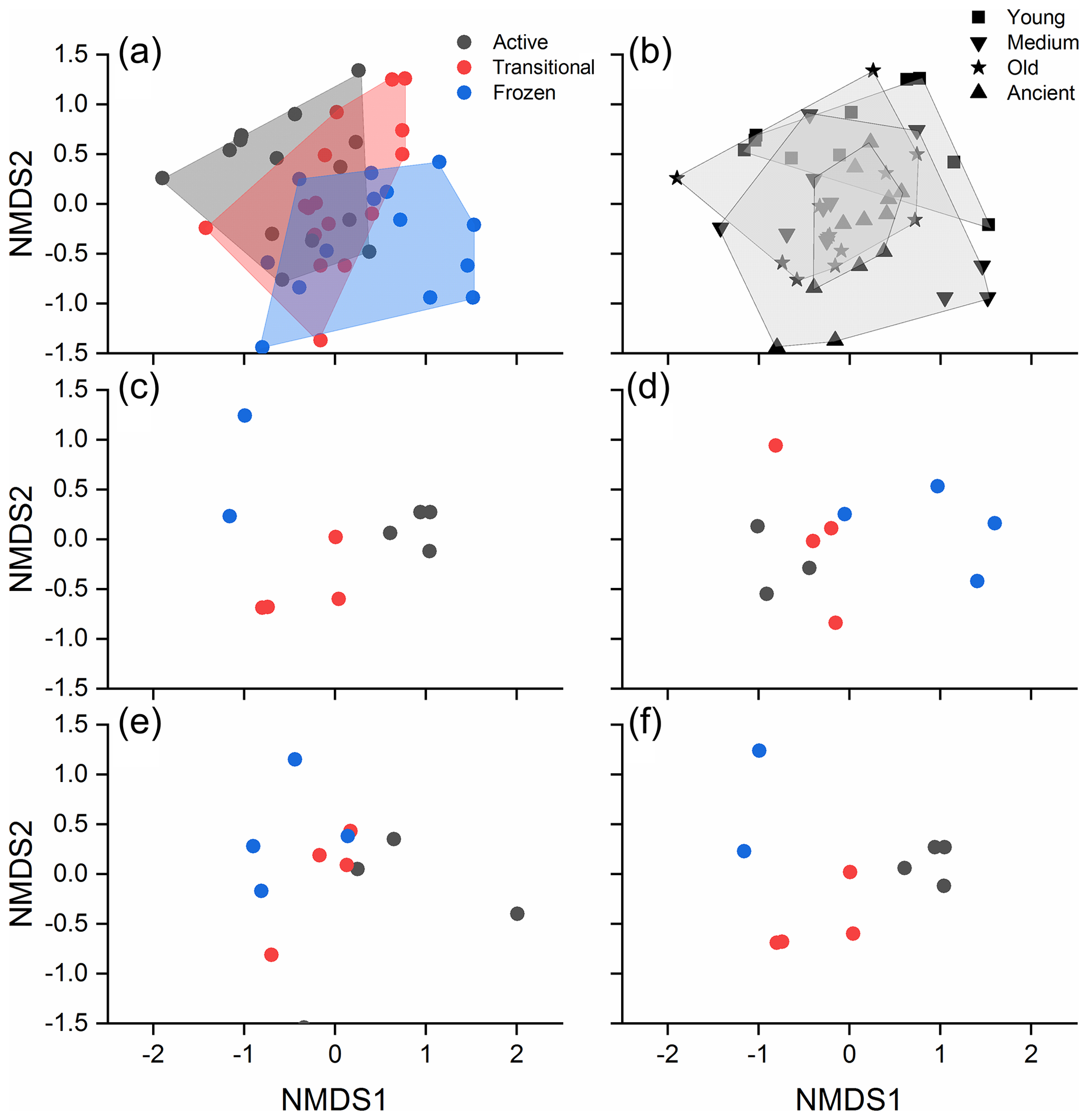
Figure 3NMDS plots showing the bacterial community structure of different thawing status (a) and permafrost age (b). The bacterial community structure of different thawing status in the young, medium-aged, old, and ancient permafrost soils are shown in (c)–(f). Active, transitional, and permanently frozen are permafrost thawing status.
PERMANOVA indicated that significantly different community structure was observed among the various permafrost thawing status and ages (both P<0.001, Table S7), and an interactive effect of the two existed (P<0.001). Post hoc analysis indicated that the community structure differences were significantly different among the soils with distinct thawing statuses (all P<0.01, Table S8). In contrast, significant differences were only detected between the young and older permafrost soils (all P<0.05, Table S9), and between the medium-aged and ancient soils (P=0.024). PERMDISP analysis indicated that the community homogeneity was not significantly different across the different permafrost thawing status (F(2, 42) = 0.193, P=0.831). A gradual transition of bacterial community structure due to permafrost thawing was observed in each permafrost age category (Fig. 3c–f). Significantly different soil bacterial community structure across the various thawing statuses was detected in the young, medium-aged, and ancient permafrost (PERMANOVA, P=0.002, 0.027, and 0.016, respectively, Table S10), but not in the old permafrost (P=0.124). Similarly, significantly different soil bacterial structure was also detected among the permafrost of different ages with the same thawing status (Table S11; Fig. S3).
3.4 The influence of permafrost thawing status and age on the community assembly of bacteria
The average contribution of stochasticity to community assembly was 68 %, 74 %, and 86 % in the active, transitional, and frozen layers of the permafrost. Significant differences in the contribution of stochasticity were detected between the active and frozen layer and between the transitional and frozen layers (both P<0.05; Fig. S4a), but not between the active and transitional layers (P=0.15). In contrast, the average contribution of stochasticity was 65 %, 76 %, 68 %, and 76 % for the young, medium-aged, old, and ancient permafrost, with no significant contribution differences among the different ages of permafrost (all P>0.05; Fig. S4b).
3.5 Quantifying the influence of permafrost thawing status and age on bacterial richness and community structure variation
Structural equation modeling (SEM) revealed that both permafrost thawing status and age significantly contributed to bacterial richness. Permafrost thawing status exhibited a higher contribution than age (standard regression weight of 0.51 and −0.30, respectively, both P<0.05) to bacterial richness (Fig. 4a). However, the influences of TOC and TN on bacterial richness were not detected. This is consistent with the random forest analysis results, which only identified permafrost thawing and age as the significant determinants of bacterial richness (Fig. S5). For community structure, permafrost thawing exhibited an indirect influence on NMDS1 via TN (standard regression weight of 0.58 and −0.63, both P<0.001; Fig. 4b). In comparison, both permafrost age and thawing status significantly contributed to NMDS2 (standard regression weight of −0.34 and 0.59, respectively, both P<0.01), while TN also exhibited a significant influence on NMDS2 (−0.49, P=0.002). The significant contributions of TN, permafrost thawing, and age were consistently identified using the random forest approach (Fig. S5).
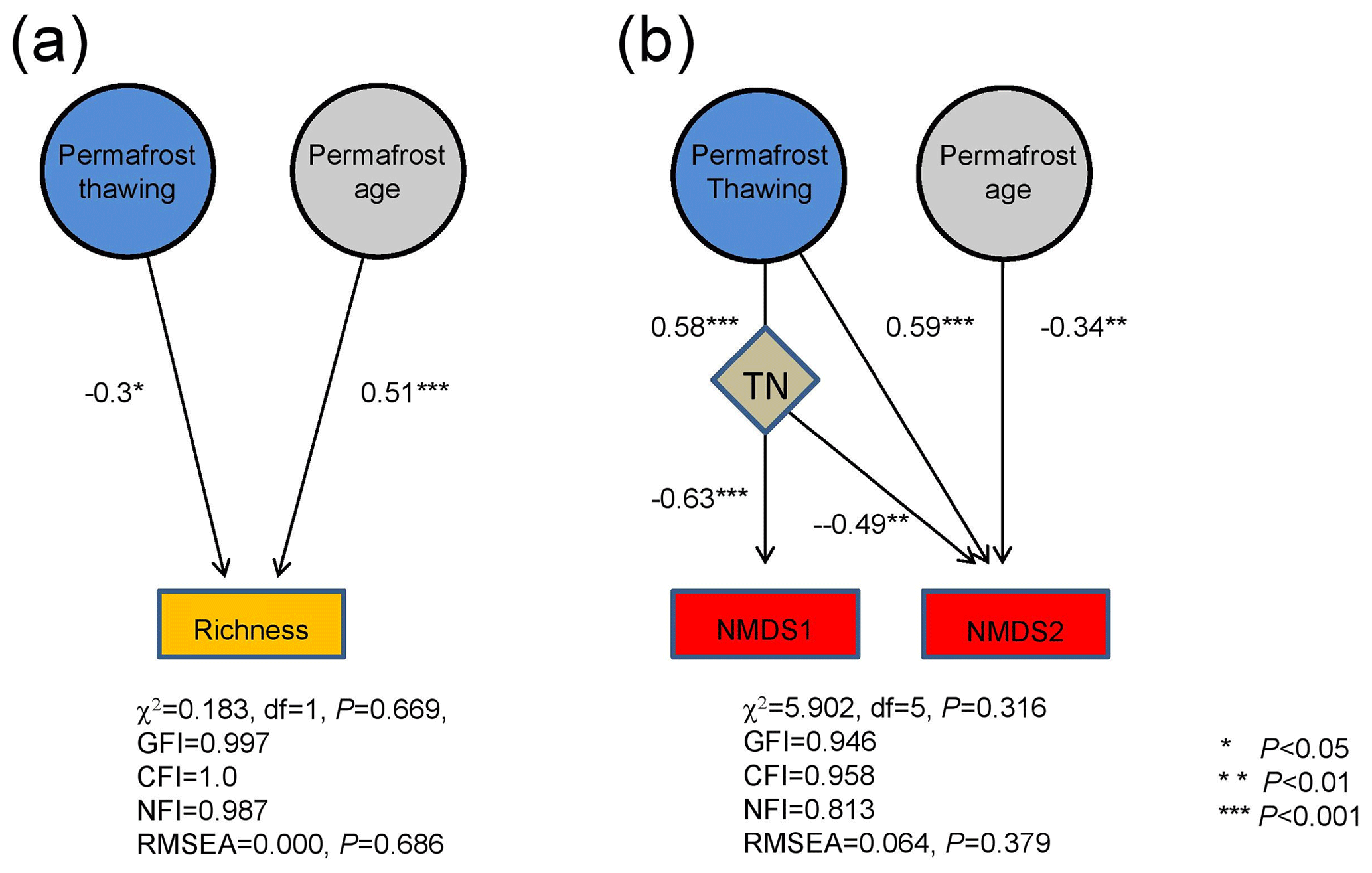
Figure 4The relative importance of permafrost thawing status and age on bacterial richness (a) and community structure (b) based on structural equation modeling. The community structure variation was assessed by the first and second axis coordinates of the NMDS plot (NMDS1 and NMDS2). Numbers adjacent to arrows are the absolute values of the path coefficients, indicative of the standardized effect size of the relationship. ∗: P<0.05, : P<.01, and : P<0.001.
The bacteria richness was significantly higher in the active layer soil (Fig. 2a), and this is consistent with the previous findings that permafrost thawing significantly increased bacterial richness in soil in the Tibetan Plateau and the high Arctic (Chen et al., 2017; Schostag et al., 2019; Wu et al., 2018). Permafrost thawing leads to accelerated microbial degradation of soil organic carbon that can generate a wide variety of metabolic products (Mueller et al., 2015). The increased metabolic product diversity would lead an increased nutrient diversity and provide additional ecological niches for bacteria (Hernández and Hobbie, 2010). This would explain the increased bacterial richness observed in our study. Furthermore, the phylogenetic diversity exhibited a greater sensitivity to permafrost thawing than the Shannon diversity (Fig. S2). As phylogenetically close-related microorganisms have similar habitat associations, phylogeny-based community metrics could infer potential community functional change (Stegen et al., 2012). Hence, this suggests that community function could be more sensitive to permafrost thawing than community composition.
The soil bacteria in the young permafrost exhibited a stronger response to thawing than those in older permafrost soils (Fig. 2b). The young permafrost soil demonstrated a higher relative abundance of aliphatic carbon but lower carbohydrates than older permafrost soils (Mueller et al., 2015). Thus, bacterial richness could be driven by carbon quality, but not quantity. It has been reported that the degradation of complex carbon molecules requires extensive microbial collaboration, thus leading to a more diverse microbial community in forest soil (Ding et al., 2015). Furthermore, an earlier study on the freshwater ecosystem also confirmed that organic carbon composition determined bacterial richness and community structure (Docherty et al., 2006). This is in agreement with the higher bacterial richness detected in the active layer of the young permafrost soil (Fig. 2b).
The increased bacterial richness due to permafrost thawing was mainly attributed to Firmicutes, Actinobacteria, Chloroflexi, Deltaproteobacteria, and Alphaproteobacteria in the young permafrost soil (Table S6). An increased transcriptional response of Chloroflexi has been reported during permafrost thawing (Coolen and Orsi, 2015) and may be attributed to their recalcitrate organic matter degradation capacity (Colatriano et al., 2018). Firmicutes and Actinobacteria have been reported to be more abundant in the frozen layer than in the active layer of permafrost soil due to their capacities in maintaining metabolic activity and DNA repair mechanisms at low temperatures (Johnson et al., 2007; Tuorto et al., 2014). However, our results showed that their diversity may increase during permafrost thawing, despite their reduction in relative abundance. Alphaproteobacteria and Deltaproteobacteria were both abundant in the upper permafrost soil in the Tibetan Plateau, and their relative abundance negatively correlated with soil depth (Wu et al., 2017). Alphaproteobacteria was identified to be more abundant in the active layer of the permafrost soil in Norway (Mueller et al., 2018). One possible explanation is that the surface active layer may be the major location for root exudates, which favors Alphaproteobacteria (Morgalev et al., 2017). Deltaproteobacteria has been reported to have strong catabolic potentials on recalcitrate aromatic compounds and plant detritus (Jansson and Tas, 2014). This may explain the enhanced richness of Deltaproteobacteria in the active layer of permafrost.
PERMANOVA, SEM, and random forest analyses consistently demonstrated statistically significant contributions of permafrost thawing and age to soil bacterial community structure (Fig. 4b). Bacterial communities were better separated by thawing status than by age on the NMDS plots (Fig. 3a and b). Furthermore, a significantly higher contribution of determinism (lower stochasticity) was observed in the thawed permafrost soils (active and transitional layers) than in the permanently frozen layer. This is contrastively different from the weak influence of permafrost age on the bacterial community (Fig. S4). Collectively, these suggest that permafrost thawing has a stronger influence on bacterial community structure than permafrost age. Our results are consistent with Mondav et al. (2017), who found that permafrost activity separated the community structure better than soil depth in peatland permafrost soil in Sweden.
Permafrost thawing significantly increased determinism in bacterial community structure (Fig. S4). Increased determinism is frequently attributed to enhanced environmental filtering (Stegen et al., 2012). Our results demonstrated that TN and the C:N ratio explained a greater proportion of the bacterial community structure than TOC. This is consistent with the previous findings that nitrogen availability strongly regulates microbial community structure and function in the permafrost soils of the Arctic and Tibetan Plateau (Chen et al., 2018, 2017; Yergeau et al., 2010). Significantly different soil carbon and nitrogen were observed among the various permafrost thawing statuses, but not among the different permafrost ages (Fig. 1a and c). Thus the changed nutrients may explain the significant influence of thawing status on the community structure and assembly processes. The community structure change due to permafrost thawing has also been proposed to be due to the colonization of microorganisms in the active layer (Monteux et al., 2018), which coincides with the increased bacterial richness observed here (Fig. 2a).
Bacterial community structure in the active layer is more similar to the transitional layer than to the permanently frozen layer (Fig. 3). This is consistent with those observed in other Arctic permafrost settings (Monteux et al., 2018; Deng et al., 2015), confirming that thawing can homogenize bacterial community structure of different soil depths. However, significant differences in bacterial community were still observed between the active and transitional layers (Table S8), instead of being identical (Monteux et al., 2018). This could be due to physiochemical heterogeneity between the soils in the different permafrost layers (Fig. 1, Kao-Kniffin, et al., 2015; Mueller et al., 2015). Thus, variations in the measured (such as TN) and unmeasured physicochemical properties (such as pH) among the different permafrost layers also contributed to the bacterial community heterogeneity and led to the significantly different bacterial communities observed.
The influence of permafrost age on bacterial community structure was weaker (Fig. 3b), with only significantly different community structure being observed in the young and medium-aged permafrost soils (Table S8). A substantial influence of permafrost age on community structure has been reported previously (Mackelprang et al., 2017). Investigation on the pedogenesis following deglaciation also revealed distinct microbial community structure along the chronosequence (Freedman and Zak, 2015). However, the community differences between the old and ancient permafrost soils were much weaker than expected (Table S9). This is likely due to the strong influence of permafrost thawing, as thawing enhances environmental filtering (Fig. S4) and homogenizes community structure in soils of different ages. This is confirmed by the significantly different bacterial community structure in permafrost soils of the same age along the thawing gradient (except the old permafrost soil; Fig. 3c–f, Table S10).
Our results demonstrated that bacterial community structure did not converge due to permafrost thawing, as reflected by the non-significant difference in sample heterogeneity among the various permafrost layers (Fig. S3, Table S11). This contradicts previous studies (Deng et al., 2015; Yuan et al., 2018) in the Arctic but was consistent with Mackelprang (2011). Our results also contradict to Kao-Kniffin et al. (2015), which reported lowered prokaryotic community differences in the active layer than in the transitional and permanently frozen permafrost. Several reasons could cause this inconsistency. Firstly, different microbial communities were targeted. Kao-Kniffin et al. (2015) focused on the archaeal community, whereas only bacteria were targeted in the present study. Kao-Kniffin et al. (2015) identified a single archaeon OTU accounting for over 30 % of the prokaryotic community (Fig. 3 in Kao-Kniffin et al., 2015). An early study revealed that archaea exhibited a lower community variation with increasing soil depths compared with bacteria (Frank-Fahle et al., 2014). Therefore, the community convergence observed by Kao-Kniffin et al. (2015) could be due to the influence of archaea. Furthermore, the inconsistency may be related to the different community dissimilarity metrics used. Kao-Kniffin et al. (2015) used unweighted UniFrac, which only calculates the phylogenetic closeness of OTUs, and the relative abundance is not considered. This is distinctively different from the Bray–Curtis dissimilarity used in the present study, and it has been reported that unweighted and weighted community metrics examine different features of community structure (Lozupone et al., 2011).
The distinct bacterial community structure in the variously aged permafrost soils, yet under the same thawing status, confirms the historic effects of permafrost age on the community structure during permafrost thawing. The distinct bacterial community structure is likely to result in different metabolic functions (Brown and Forsythe, 1974); thus the significantly different bacterial structure under the same thawing status may lead to different organic carbon degradation capacities. Furthermore, older permafrost enriches pathways involved in the degradation of recalcitrant biomass, while decreasing pathways associated with starch and sucrose metabolisms compared with younger soils (Mackelprang et al., 2017). Thus, the thawing of permafrost soils of different ages may also lead to distinct soil carbon degradation schemes.
Our results demonstrated that permafrost thawing consistently exhibited greater influence on bacterial richness and community structure than permafrost age. However, permafrost age alters the response of permafrost soil bacteria to thawing, with a stronger response to thawing observed in the young than older permafrost soils. The different community structure during permafrost thawing may present distinct metabolic potentials for soil organic carbon cycling, and may ultimately alter the carbon emission scheme. Further studies are required to identify the environmental and historical factors that lead to the distinct responses of bacteria in the permafrost of different ages.
Sequence data generated in the present study have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive under the ID PRJNA554442.
The supplement related to this article is available online at: https://doi.org/10.5194/tc-14-3907-2020-supplement.
WK conceived the study and developed the idea with MJ. TZ and HZ performed DNA extraction. MJ conducted the data statistical analysis. MJ and WK wrote the first draft of the paper, and CL and XD revised the paper substantially. All authors read and approved the final paper.
The authors declare that they have no conflict of interest.
We greatly thank Jenny Kao-Kniffin for kindly providing the permafrost soil samples for this study. This project was financially supported by Chinese Academy of Sciences (XDA19070304, QYZDB-SSW-DQC033, and XDA20050101), and National Natural Science Foundation of China (41771303).
This research has been supported by the Chinese Academy of Sciences (grant no. XDA19070304, QYZDB-SSW-DQC033, and XDA20050101) National Natural Science Foundation of China (41771303).
This paper was edited by Christian Hauck and reviewed by two anonymous referees.
Anderson, M.: Distance–based tests for homogeneity of multivariate dispersions, Biometrics, 62, 245–253, 2006.
Anderson, M. J.: A new method for non–parametric multivariate analysis of variance, Austral Ecol., 26, 32–46, 2001.
Archer, E.: RfPermute: Version 2.1.5 to Accompany R Journal Paper, Zenodo, https://doi.org/10.5281/zenodo.159219, 2016.
Bardgett, R. D. and Walker, L. R.: Impact of coloniser plant species on the development of decomposer microbial communities following deglaciation, Soil Biol. Biochem., 36, 555–559, 2004.
Brown, M. B. and Forsythe, A. B.: Robust tests for the equality of variances, J. Am. Stat. Assoc., 69, 364–367, 1974.
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., Owens, S. M., Betley, J., Fraser, L., Bauer, M., Gormley, N., Gilbert, J. A., Smith, G., and Knight, R.: Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms, ISME J., 6, 1621–1624, 2012.
Chen, L., Liang, J., Qin, S., Liu, L., Fang, K., Xu, Y., Ding, J., Li, F., Luo, Y., and Yang, Y.: Determinants of carbon release from the active layer and permafrost deposits on the Tibetan Plateau, Nat. Commun., 7, 13046–13046, 2016.
Chen, L., Liu, L., Mao, C., Qin, S., Wang, J., Liu, F., Blagodatsky, S., Yang, G., Zhang, Q., Zhang, D., Yu, J., and Yang, Y.: Nitrogen availability regulates topsoil carbon dynamics after permafrost thaw by altering microbial metabolic efficiency, Nat. Commun., 9, 3951, https://doi.org/10.1038/s41467-018-06232-y 2018.
Chen, Y.-L., Deng, Y., Ding, J.-Z., Hu, H.-W., Xu, T.-L., Li, F., Yang, G.-B., and Yang, Y.-H.: Distinct microbial communities in the active and permafrost layers on the Tibetan Plateau, Microb. Ecol., 26, 6608–6620, 2017.
Clarke, K. R. and Warwick, R. M.: PRIMER v6: user manual/tutorial, PRIMER–E, Plymouth, 2006.
Colatriano, D., Tran, P. Q., Gueguen, C., Williams, W. J., Lovejoy, C., and Walsh, D. A.: Genomic evidence for the degradation of terrestrial organic matter by pelagic Arctic Ocean Chloroflexi bacteria, Commun. Biol., 1, 90–90, 2018.
Coolen, M. J. L. and Orsi, W. D.: The transcriptional response of microbial communities in thawing Alaskan permafrost soils, Front. Microbiol., 6, 14, https://doi.org/10.3389/fmicb.2015.00197, 2015.
Deng, J., Gu, Y., Zhang, J., Xue, K., Qin, Y., Yuan, M., Yin, H., He, Z., Wu, L., Schuur, E. A. G., Tiedje, J. M., and Zhou, J.: Shifts of tundra bacterial and archaeal communities along a permafrost thaw gradient in Alaska, Microb. Ecol., 24, 222–234, 2015.
Ding, J., Zhang, Y., Wang, M., Sun, X., Cong, J., Deng, Y., Lu, H., Yuan, T., Van Nostrand, J. D., Li, D., Zhou, J., and Yang, Y.: Soil organic matter quantity and quality shape microbial community compositions of subtropical broadleaved forests, Microb. Ecol., 24, 5175–5185, 2015.
Docherty, K. M., Young, K. C., Maurice, P. A., and Bridgham, S. D.: Dissolved organic matter concentration and quality influences upon structure and function of freshwater microbial communities, Microb. Ecol., 52, 378–388, 2006.
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R.: UCHIME improves sensitivity and speed of chimera detection, Bioinformatics, 27, 2194–2200, 2011.
Frank-Fahle, B. A., Yergeau, E., Greer, C. W., Lantuit, H., and Wagner, D.: Microbial functional potential and community composition in permafrost–affected soils of the NW Canadian Arctic, PLoS One, 9, e84761, https://doi.org/10.1371/journal.pone.0084761, 2014.
Freedman, Z. and Zak, D. R.: Soil bacterial communities are shaped by temporal and environmental filtering: evidence from a long–term chronosequence, Appl. Environ. Microbiol., 17, 3208–3218, 2015.
Gilichinskii, D. A.: Microbial life in permafrost: a historical review, Permafr. Periglac., 6, 243–250, 1995.
Guo, G., Kong, W., Liu, J., Zhao, J., Du, H., Zhang, X., and Xia, P.: Diversity and distribution of autotrophic microbial community along environmental gradients in grassland soils on the Tibetan Plateau, Appl. Microbiol. Biotechnol., 99, 8765–8776, 2015.
Hernández, D. L. and Hobbie, S. E.: The effects of substrate composition, quantity, and diversity on microbial activity, Plant Soil, 335, 397–411, 2010.
Hinkel, K. M., Eisner, W. R., Bockheim, J. G., Nelson, F. E., Peterson, K. M., and Dai, X.: Spatial extent, age, and carbon stocks in drained thaw lake basins on the Barrow peninsula, Alaska, Arct. Antarct. Alp. Res., 35, 291–300, 2003.
Hobbie, S. E., Schimel, J. P., Trumbore, S. E., and Randerson, J. R.: Controls over carbon storage and turnover in high–latitude soils, Glob. Change Biol., 6, 196–210, 2000.
Jansson, J. K. and Tas, N.: The microbial ecology of permafrost, Nat. Rev. Microbiol., 12, 414–425, 2014.
Johnson, S. S., Hebsgaard, M. B., Christensen, T. R., Mastepanov, M., Nielsen, R., Munch, K., Brand, T., Gilbert, M. T., Zuber, M. T., Bunce, M., Ronn, R., Gilichinsky, D., Froese, D., and Willerslev, E.: Ancient bacteria show evidence of DNA repair, P. Natl. Acad. Sci. USA, 104, 14401–14405, 2007.
Kao-Kniffin, J., Woodcroft, B. J., Carver, S. M., Bockheim, J. G., Handelsman, J., Tyson, G. W., Hinkel, K. M., and Mueller, C. W.: Archaeal and bacterial communities across a chronosequence of drained lake basins in arctic alaska, Sci. Rep., 5, 18165, https://doi.org/10.1038/srep18165, 2015.
Kazemi, S., Hatam, I., and Lanoil, B.: Bacterial community succession in a high–altitude subarctic glacier foreland is a three–stage process, Mol. Ecol., 25, 5557–5567, 2016.
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., Blomberg, S. P., and Webb, C. O.: Picante: R tools for integrating phylogenies and ecology, Bioinformatics, 26, 1463–1464, 2010.
Kim, M., Jung, J. Y., Laffly, D., Kwon, H. Y., and Lee, Y. K.: Shifts in bacterial community structure during succession in a glacier foreland of the High Arctic, FEMS Microbiol. Ecol., 93, 9, https://doi.org/10.1093/femsec/fiw213, 2017.
Kwon, H. Y., Jung, J. Y., Laffly, D., Lim, H. S., and Lee, Y. K.: Soil development and bacterial community shifts along the chronosequence of the Midtre Lovenbreen glacier foreland in Svalbard, J. Ecol. Nat. Environ., 38, 461–476, 2015.
Liu, J., Kong, W., Zhang, G., Khan, A., Guo, G., Zhu, C., Wei, X., Kang, S., and Morgan–Kiss, R. M.: Diversity and succession of autotrophic microbial community in high–elevation soils along deglaciation chronosequence, FEMS Microbiol. Ecol., 92, fiw160, https://doi.org/10.1093/femsec/fiw160, 2016.
Lozupone, C., Lladser, M., Knights, D., Stombaugh, J., Knight, R.: UniFrac: an effective distance metric for microbial community comparison, ISME J., 5, 169–172, 2011.
Mackelprang, R., Waldrop, M. P., DeAngelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., Rubin, E. M., and Jansson, J. K.: Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw, Nature, 480, 368–371, 2011.
Mackelprang, R., Burkert, A., Haw, M., Mahendrarajah, T., Conaway, C. H., Douglas, T. A., and Waldrop, M. P.: Microbial survival strategies in ancient permafrost: insights from metagenomics, ISME J., 11, 2305–2318, 2017.
Mondav, R., McCalley, C. K., Hodgkins, S. B., Frolking, S., Saleska, S. R., Rich, V. I., Chanton, J. P., and Crill, P. M.: Microbial network, phylogenetic diversity and community membership in the active layer across a permafrost thaw gradient, Appl. Environ. Microbiol., 19, 3201–3218, 2017.
Monteux, S., Weedon, J. T., Blume–Werry, G., Gavazov, K., Jassey, V. E. J., Johansson, M., Keuper, F., Olid, C., and Dorrepaal, E.: Long–term in situ permafrost thaw effects on bacterial communities and potential aerobic respiration, ISME J., 12, 2129–2141, 2018.
Morgalev, Y. N., Lushchaeva, I. V., Morgaleva, T. G., Kolesnichenko, L. G., Loiko, S. V., Krickov, I. V., Lim, A., Raudina, T. V., Volkova, I. I., Shirokova, L. S., Morgalev, S. Y., Vorobyev, S. N., Kirpotin, S. N., and Pokrovsky, O. S.: Bacteria primarily metabolize at the active layer/permafrost border in the peat core from a permafrost region in western Siberia, Polar Bio., 40, 1645–1659, 2017.
Mueller, C. W., Rethemeyer, J., Kao-Kniffin, J., Loeppmann, S., Hinkel, K. M., and G. Bockheim, J.: Large amounts of labile organic carbon in permafrost soils of northern Alaska, Glob. Change Biol., 21, 2804–2817, 2015.
Mueller, O., Bang–Andreasen, T., White III, R. A., Elberling, B., Tas, N., Kneafsey, T., Jansson, J. K., and Ovreas, L.: Disentangling the complexity of permafrost soil by using high resolution profiling of microbial community composition, key functions and respiration rates, Appl. Environ. Microbiol., 20, 4328–4342, 2018.
Ning, D. L., Deng, Y., Tiedje, J. M., and Zhou, J. Z.: A general framework for quantitatively assessing ecological stochasticity, P. Natl. Acad. Sci. USA, 116, 16892–16898, 2019.
Park, S. J., Park, B. J., Jung, M. Y., Kim, S. J., Chae, J. C., Roh, Y., Forwick, M., Yoon, H. I., and Rhee, S. K.: Influence of deglaciation on microbial communities in marine sediments off the coast of Svalbard, Arctic circle, Microb. Ecol., 62, 537–548, 2011.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., and Robinson, C. J.: Introducing mothur: open–source, platform–Independent, community–supported software for describing and comparing microbial communities, Appl. Environ. Microbiol., 75, 7537–7541, 2009.
Schostag, M., Prieme, A., Jacquiod, S., Russel, J., Ekelund, F., and Jacobsen, C. S.: Bacterial and protozoan dynamics upon thawing and freezing of an active layer permafrost soil, ISME J., 13, 1345–1359, 2019.
Stegen, J. C., Lin, X., Konopka, A. E., and Fredrickson, J. K.: Stochastic and deterministic assembly processes in subsurface microbial communities, ISME J., 6, 1653–1664, 2012.
Tuorto, S. J., Darias, P., McGuinness, L. R., Panikov, N., Zhang, T., Haeggblom, M. M., and Kerkhof, L. J.: Bacterial genome replication at subzero temperatures in permafrost, ISME J., 8, 139–149, 2014.
Turetsky, M. R., Abbott, B. W., Jones, M. C., Anthony, K. W., Olefeldt, D., Schuur, E. A. G., Koven, C., McGuire, A. D., Grosse, G., Kuhry, P., Hugelius, G., Lawrence, D. M., Gibson, C., and Sannel, A. B. K.: Permafrost collapse is accelerating carbon release, Nature, 569, 32–34, 2019.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R.: Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy, Appl. Environ. Microbiol., 73, 5261–5267, 2007.
Wu, X., Xu, H., Liu, G., Ma, X., Mu, C., and Zhao, L.: Bacterial communities in the upper soil layers in the permafrost regions on the Qinghai–Tibetan plateau, Appl. Soil Ecol., 120, 81–88, 2017.
Wu, X., Xu, H., Liu, G., Zhao, L., and Mu, C.: Effects of permafrost collapse on soil bacterial communities in a wet meadow on the northern Qinghai–Tibetan Plateau, BMC Ecol., 18, 27 https://doi.org/10.1186/s12898-018-0183-y, 2018.
Yang, Y. H., Fang, J. Y., Smith, P., Tang, Y. H., Chen, A. P., Ji, C. J., Hu, H. F., Rao, S., Tan, K., and He, J. S.: Changes in topsoil carbon stock in the Tibetan grasslands between the 1980s and 2004, Glob. Change Biol., 15, 2723–2729, 2009.
Yergeau, E., Hogues, H., Whyte, L. G., and Greer, C. W.: The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses, ISME J., 4, 1206–1214, 2010.
Yuan, M. M., Zhang, J., Xue, K., Wu, L., Deng, Y., Deng, J., Hale, L., Zhou, X., He, Z., Yang, Y., Van Nostrand, J. D., Schuur, E. A. G., Konstantinidis, K. T., Penton, C. R., Cole, J. R., Tiedje, J. M., Luo, Y., and Zhou, J.: Microbial functional diversity covaries with permafrost thaw–induced environmental heterogeneity in tundra soil, Glob. Change Biol., 24, 297–307, 2018.