the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 23 Jan 2020
| 23 Jan 2020
Impact of exhaust emissions on chemical snowpack composition at Concordia Station, Antarctica
Detlev Helmig
Daniel Liptzin
Jacques Hueber
Joel Savarino
The chemistry of reactive gases inside the snowpack and in the lower atmosphere was investigated at Concordia Station (Dome C), Antarctica, from December 2012 to January 2014. Measured species included ozone, nitrogen oxides, gaseous elemental mercury (GEM), and formaldehyde, for study of photochemical reactions, surface exchange, and the seasonal cycles and atmospheric chemistry of these gases. The experiment was installed ≈1 km from the station main infrastructure inside the station clean air sector and within the station electrical power grid boundary. Ambient air was sampled continuously from inlets mounted above the surface on a 10 m meteorological tower. In addition, snowpack air was collected at 30 cm intervals to 1.2 m depth from two manifolds that had both above- and below-surface sampling inlets. Despite being in the clean air sector, over the course of the 1.2-year study, we observed on the order of 50 occasions when exhaust plumes from the camp, most notably from the power generation system, were transported to the study site. Continuous monitoring of nitrogen oxides (NOx) provided a measurement of a chemical tracer for exhaust plumes. Highly elevated levels of NOx (up to 1000 × background) and lowered ozone (down to ≈50 %), most likely from reaction of ozone with nitric oxide, were measured in air from above and within the snowpack. Within 5–15 min from observing elevated pollutant levels above the snow, rapidly increasing and long-lasting concentration enhancements were measured in snowpack air. While pollution events typically lasted only a few minutes to an hour above the snow surface, elevated NOx levels were observed in the snowpack lasting from a few days to ≈ 1 week. GEM and formaldehyde measurements were less sensitive and covered a shorter measurement period; neither of these species' data showed noticeable concentration changes during these events that were above the normal variability seen in the data. Nonetheless, the clarity of the NOx and ozone observations adds important new insight into the discussion of if and how snow photochemical experiments within reach of the power grid of polar research sites are possibly compromised by the snowpack being chemically influenced (contaminated) by gaseous and particulate emissions from the research camp activities. This question is critical for evaluating if snowpack trace chemical measurements from within the camp boundaries are representative for the vast polar ice sheets.
- Article
(4668 KB) - Full-text XML
-
Supplement
(576 KB) - BibTeX
- EndNote





Research conducted during the past ≈15 years has revealed an active and remarkable spatial diversity of atmospheric oxidation chemistry in the polar lower atmosphere (Grannas et al., 2007). Ozone plays a fundamental role in controlling the lifetime of many atmospheric trace gases directly and indirectly by modulating atmospheric OH. Unlike the episodic ozone depletion events observed at coastal sites, the opposite effect (i.e., ozone production) has been observed in the Antarctic interior (Crawford et al., 2001; Helmig et al., 2007b, 2008a; Legrand et al., 2009, 2016). The discovery of ozone production chemistry in the remote and pristine Antarctic environment was rather surprising because hitherto photochemical production in the lower atmosphere had exclusively been associated with polluted urban environments (Molina and Molina, 2004). Photochemical production and snowpack emissions of nitric oxides (NOx) have been identified as underlying processes driving this chemistry. NOx has been shown to be formed from photochemical reactions in the snowpack (Honrath et al., 1999; Jones et al., 2000), with deposited nitrate constituting the reservoir of this chemistry. NOx plays a crucial role in snow photochemical reactivity (Murray et al., 2015). NOx mixing ratios in interstitial air resulting from photochemical reactions can exceed those in the air above the snowpack by a factor of ≈50 (Van Dam et al., 2015).
This concentration gradient is driving NOx emission fluxes out of the snowpack into the overlying atmosphere (Jones et al., 2001; Honrath et al., 2002), which, under stable atmospheric conditions, can cause large NOx enhancements in the atmospheric surface layer (Helmig et al., 2008b; Neff et al., 2008; Frey et al., 2011, 2013), and in the presence of solar irradiance trigger photochemical ozone production, with resulting peak ozone levels that can be double those in the atmospheric layer aloft (Crawford et al., 2001; Helmig et al., 2008a; Legrand et al., 2016). Experiments on reactive nitrogen chemistry investigating this rather unexpected ozone production chemistry have built on a variety of atmospheric research strategies, including snowpack air sampling (Dibb et al., 2002; Jacobi et al., 2004; Helmig et al., 2007a; Van Dam et al., 2015), snow flux chambers (Dibb et al., 2002), snow chemical analyses (Dassau et al., 2002; Dibb et al., 2007b; France et al., 2011; Erbland et al., 2013), atmospheric monitoring (Frey et al., 2011; Kramer et al., 2015; Legrand et al., 2016), surface fluxes (Jones et al., 2001; Honrath et al., 2002; Frey et al., 2011, 2015), and boundary layer vertical profiling (Helmig et al., 2008a; Frey et al., 2015).
Most of these studies have relied on observations from dedicated campaigns at research stations, including photochemistry campaigns at Summit, Greenland (Dibb et al., 2007a), the Antarctic Tropospheric Chemistry Investigation (ANTCI; Eisele and Davis, 2008) at the South Pole, the Chemistry of the Antarctic Boundary Layer and the Interface with Snow (CHABLIS) experiment at Halley (Jones et al., 2008), and the Oxidant Production over Antarctic Land and its Export (OPALE) campaign at Concordia Station (Preunkert et al., 2012). A common limitation of these studies is that experiments were conducted in proximity to research stations, where use of fuel-powered engines in generators and vehicles causes exhaust emissions with highly elevated concentrations of particulates and gases, particularly of volatile organic compounds (VOCs) and NOx. A critical question is if and how this pollution, and possibly secondary products formed during the atmospheric transport and deposition, impacts the snow chemical position and reactivity, and potentially the findings from this aforementioned literature. This is of particular importance for oxidized nitrogen species.
This study yielded, for the first time, a year-long record of NOx and O3 in an Antarctic snowpack at Concordia and the atmosphere above it. This experiment also gave us the opportunity to study and evaluate occurrences of pollution episodes, using the NOx monitoring as a sensitive chemical tracer for identification of exhaust plumes.
Location
This experiment was conducted at the French–Italian Antarctic research station Concordia, located at the Dome Circe or Dome Charlie (Dome C, 75.10∘ S, 123.35∘ E, 3233 m a.s.l., mean temperature – 55 ∘C). An experimental site was established at the border of the clean air sector, approximately 1 km to the west of the station common buildings (Fig. 1). The clean air sector is located in the opposite direction of the prevailing wind direction. The site consisted of a underground laboratory positioned at the border of the clean air area, a 10 m tall meteorological tower, and two snow air sampling manifolds for sampling the atmosphere and the snow interstitial air (Fig. 2). The installation was in late November 2012 with continuous monitoring conducted until January 2014 (14 months).
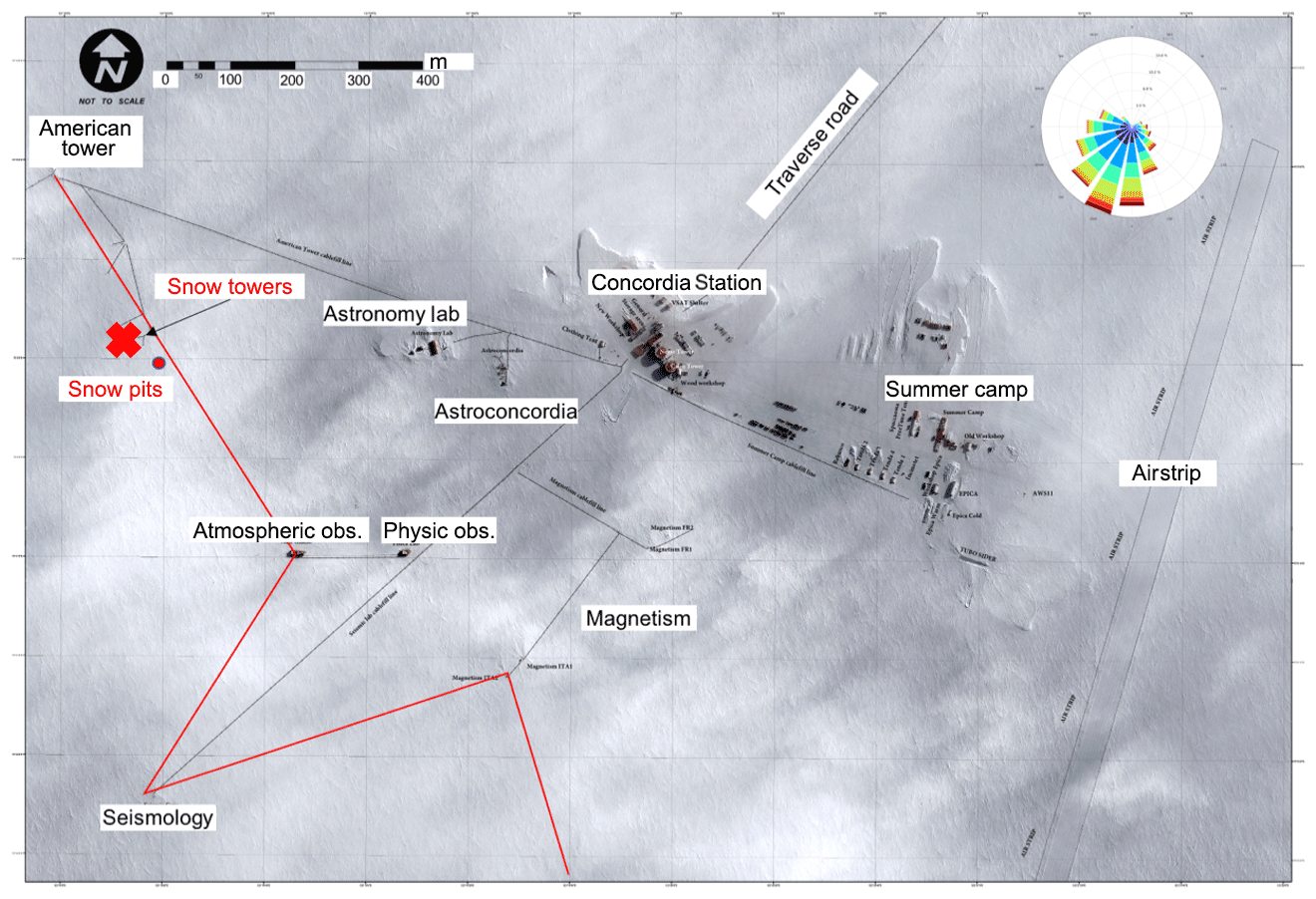
Figure 1Satellite image of research station Concordia with location of the snow photochemistry experiment indicated by the red text and cross marker. Its location was ∼1 km west of the station main buildings and power generation plant. The map also indicates the location where the snow pits were collected, the border of the clean air sector (which is west of the red line), and a wind rose for a full year of wind data collected from the met tower that was adjacent to the snow tower manifolds (Pléiades satellite image courtesy of Airbus Defence and Space SAS, CNES, 2016).

Figure 2(a) Meteorological tower with the station infrastructure in the background. The wooden box to the left is the entry hatch to the underground laboratory. Air sampling inlets were located at 0.5, 2, and 10 m above the surface on the tower. Two snowpack air sampling towers were located approximately 7 m to the left and 10 m to the right of the base of the meteorological tower. (b) One of the two snowpack air sampling manifolds (snow tower), with one pair of inlets right on the snow surface and one inlet pair at ∼30 cm height. Four more equivalent sampling inlet pairs are below the snow surface at 30 cm depth intervals extending to a maximum depth of 1.2 m (see Fig. 1 in Van Dam et al., 2015, for a schematic of a similar installation at Summit, Greenland).
Meteorological tower
A 10 m meteorological tower (Fig. 2a) was equipped with two sonic anemometers for atmospheric turbulence measurements and three gas sampling inlets (0.5, 2, 10 m) with sampling lines inside a heated conduit running to the laboratory. The upper inlet was attached to a manual pulley allowing it to be lowered for side-by-side sampling of both inlets for tracking and correcting sampling inlet and line biases.
Snowpack air sampling
Two identical multi-inlet snow sampling manifolds (“snow tower”) for collection of interstitial and ambient air were constructed, with a design similar to that described by Seok et al. (2009) (Fig. 2b). The snow tower consisted of a vertical post of square aluminum alloy (3.8 cm × 3.8 cm) with eight 60 cm long cross arms spaced vertically every 20 cm. Each of the cross bars supported a pair of sampling inlets. The inlets were fitted with 25 mm Acrodisc hydrophobic polytetrafluoroethylene (PTFE) syringe filters (Pall Life Sciences, Ann Arbor, Michigan, USA) to prevent snow and ice crystals from being pulled into the sampling line. For the installation, a snow pit was dug and the inlets were inserted horizontally into the clean untouched walls of the pit. The hole was then loosely refilled with the excavated snow, re-establishing the stratigraphy as much as possible. Blowing snow then refilled any remaining gaps within the following 2–3 d. The snow tower was kept in place after the campaign, so we have no data on the potential changes in porosity and air flow dynamics that resulted from the installation and subsequent changes in the snow morphology as the snowpack re-equilibrated. Insulated and heated sampling lines connected the sampling inlets to the chemical analyzers in the underground laboratory. All sampling lines were of 0.64 cm o.d. × 30 m long pre-conditioned perfluroalkoxy (PFA) tubing, except the lines to the gradient inlets on the meteorological tower, which were of 0.78 cm o.d. These were continuously purged to maintain a flow of at least 2 L min−1. Air was pulled through the snow tower sampling lines by the combined flow of the gas analyzers (ozone monitor at 1 L min−1, a gaseous elemental mercury (GEM) analyzer at 1 L min−1, NOx monitor at 1 L min−1). Only two monitors sampled from the snow tower inlets together at a given time to limit the maximum snow air sampling. Since each line connected to a pair of inlets at equal height, the effective flow through each inlet was ≈ 1 L min−1. Each height was sampled for 10 min every 2 h, resulting in the approximate volume of a sphere with a radius of 25 cm around each inlet every 2 h. Sampling from the two snow towers was alternated every 24 h. Each sampling inlet had a thermocouple wire attached for monitoring of the snowpack temperature gradient.
Ozone measurements
Ozone was measured with a Thermo Environmental (TEI) 49i UV absorption monitor that was calibrated against a NOAA Global Monitoring Division reference standard before field shipment.
NOx measurements
Nitrogen oxides were monitored with a TEI chemiluminescence analyzer (TEI 42C-TL). The TEI 42C-TL has two channels. The first channel measures NO via NO + O3 chemiluminescence. The second channel measures total nitrogen oxides (NOx= NO + NO2) by redirecting air through a heated (325 ∘C) molybdenum converter, which causes NO2 – including other oxidized nitrogen compounds – to be converted to NO. NO2 is then determined by subtracting NO, obtained from the first channel, from the resulting NOx signal. There are a number of other oxidized nitrogen species that can contribute to the NO2 measurement (Steinbacher et al., 2007). The error in the NO2 measurement increases with rising levels of interfering gases such as nitrous acid (HONO), peroxyacetyl nitrate (PAN), and alkyl nitrates that contribute to the NO2-mode signal. Consequently, NO2 concentrations obtained with the TEI 42C-TL represent an estimate for the sum of these oxidized nitrogen species. Field calibrations were conducted with a NIST-traceable 1 ppm NO in N2 gas standard (Scott-Marrin, Inc., Riverside, CA, USA) that was dynamically diluted to low-parts-per-billion mixing ratios. We did not bring a zero-air compressed gas cylinder to Concordia. Instead, a low-NOx dilution gas was prepared by pumping ambient air (which had significantly lower NOx levels than snowpack air) through a cartridge filled with ≈1 dm3 of granular chemisorbent (Purafil, Doraville, GA). Calibration ranges were from 0.1 to 25 ppb, and the instrument response was linear within this range. Intercept values of the linear regression, and zero values from sampling of the scrubbed air, were below 0.1 ppb.
Formaldehyde measurements
Formaldehyde was measured with a commercial analyzer by liquid fluorimetry. Details of the measurement, instrument characterization, and deployment at Concordia Station have been provided by Preunkert et al. (2015).
Gaseous elemental mercury measurements
GEM was measured with a commercial Tekran model 2537 instrument. The measurement protocol and calibration and instrument characterization details are available in Angot et al. (2016).
Snow sampling and determination
The snow pit data stem from sampling that was done at and near Concordia between January 2009 and December 2010, and at ≈3 m distance from snow tower 2 in January 2014. Snow was collected in pre-cleaned 50 mL centrifuge tubes inserted directly on a newly scraped wall of a snow pit. Nitrate concentration in snow samples was measured directly in the field, at the wet chemistry laboratory of Concordia Station. Each sample was melted at room temperatures and concentrations were determined using a colorimetric method employed routinely at Concordia (Frey et al., 2009).

Figure 3Photograph of Concordia Station illustrating the dispersion of the exhaust plume from the electrical power generating plant during conditions with a strong surface temperature inversion. The plume dissipated toward the west in the direction of the experimental site. This is a typical situation for a contamination event.
Results of the year-round snowpack and ambient monitoring, including interpretations on photochemistry, will be presented elsewhere (Helmig et al., 2020). Here, we primarily focus on occurrences of pollution transport to the sampling site and its penetration into the snowpack.
Figure 3 shows a photograph of the station main buildings. The power plant is adjacent to the two-column structure. Approximately 300 m3 of Special Antarctic Blend (SAB) diesel fuel are burned in the plant for electricity and heat generation per year. The exhaust plume from the 5 m high stack of the power plant can be seen in the picture, blowing towards the west. Due to the typical strong stratification and stability of the atmosphere near the surface, the plume does not rise far above the stack height, but instead gets transported horizontally at a height of ≈5–10 m above the snow surface. This is a typical exhaust plume dispersion behavior for a cold-region environment, seen at many other polar research stations. The plume typically does not hit the surface within the immediate distance of the stack location. Depending on the actual turbulent mixing conditions, it may take several hundred meters before the stack emissions are encountered right at the surface.
Of the gases monitored in this experiment, NOx was the most sensitive tracer for pollution impact. We chose to concentrate on NOx as the total of NO + NO2, as this is a more representative indicator for the total amount of oxidized nitrogen, whereas NO would only indicate a fraction. Further, the fractionation between NO and NOx is sensitive to other gases (such as ozone) and residence time and snowpack depth. Therefore, interpretation of NO data is more ambiguous. NOx in ambient air at Concordia remained well below 1 ppb during background conditions year-round (Helmig et al., 2020), in agreement with observations from prior shorter campaign NOx measurement at Concordia (Frey et al., 2011, 2013, 2015). We only observed elevated NO levels during pollution events. There were no elevated NO conditions that could be traced to snowpack emissions, similar to what has been reported from the South Pole (Helmig et al., 2008b; Neff et al., 2008). During pollution events, the 1 ppb threshold was exceeded very quickly in measurements taken from the meteorological tower and from above-surface snow tower inlets, with resulting NOx mixing ratios rising to as high as close to 200 ppb, representing an up to 1000-fold enhancement over background conditions (Fig. 4).
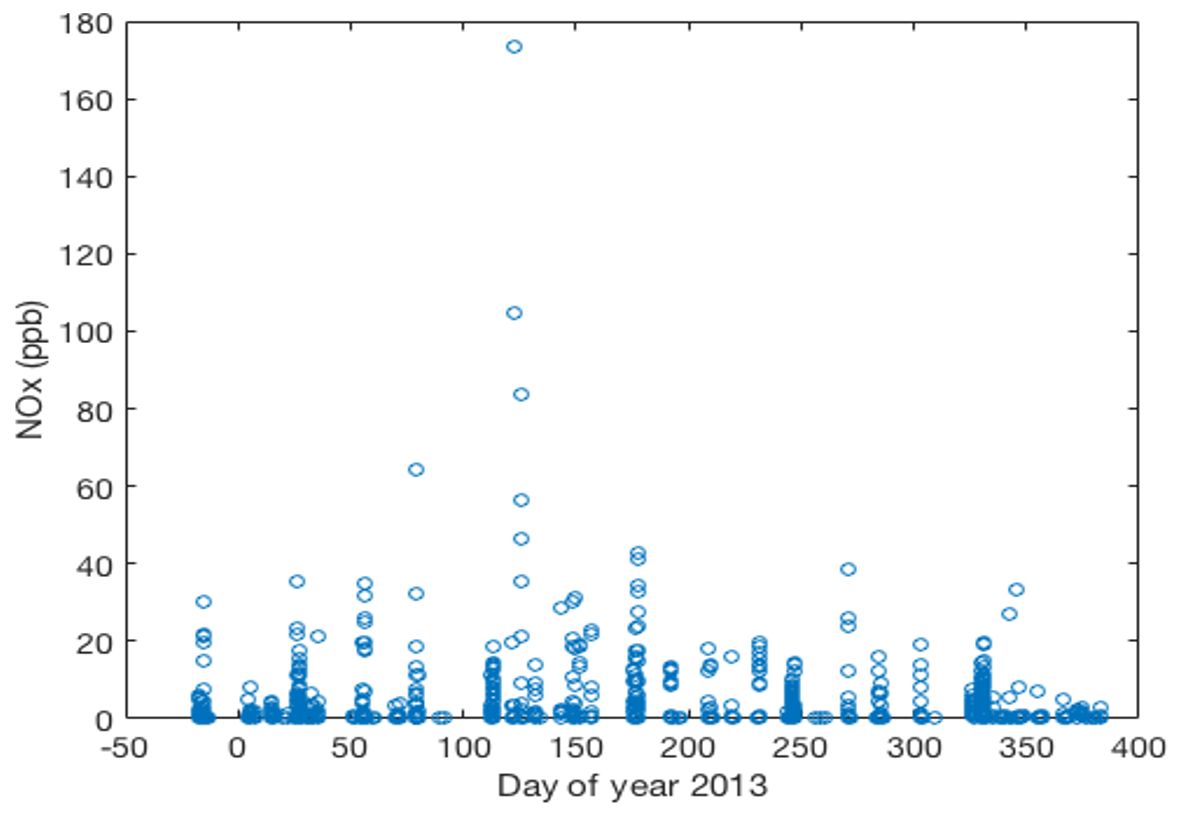
Figure 4NOx measured from the above-surface inlets on the two snow towers and from all inlets on the meteorological tower. Circles represent 10 min averaged data. Plotted are data that were extracted from occurrences when NOx was above 1 ppb, well above the background mixing ratio, and any time when the wind direction was from the polluted wind sector.
During the course of the ≈14 month study, a total of ≈50 pollution events were observed, although some events overlapped (Fig. 4). Most of these occurrences were relatively short, with elevated mixing ratios above the snowpack lasting from minutes to a few hours. We counted 15 events in total when there was a spike in the above-surface NOx measurements followed by an increase of at least 1 ppb of NOx in snowpack air. It took up to in excess of 7 d for NOx in the snowpack air to return to pre-event levels. Integrated over the entire campaign, pollution episodes constituted <2.0 % of the measurements above the snowpack and <10 % of the measurements within the snowpack. The correlation analyses of pollution occurrences with wind direction clearly define the direction of the transport. The predominant wind direction sector at Concordia is southeast to northwest (Fig. 5a), with southeasterly winds having the overall largest share. NOx levels were consistently well below 1 ppb when winds were from the southeast to northeast. The sector with pollution transport is well defined, with wind directions covering approximately 45–120∘ (Fig. 5b). These sectors perfectly line up with the upwind direction of the station power plant (Fig. 1), clearly identifying the plant as the source of these pollution occurrences.
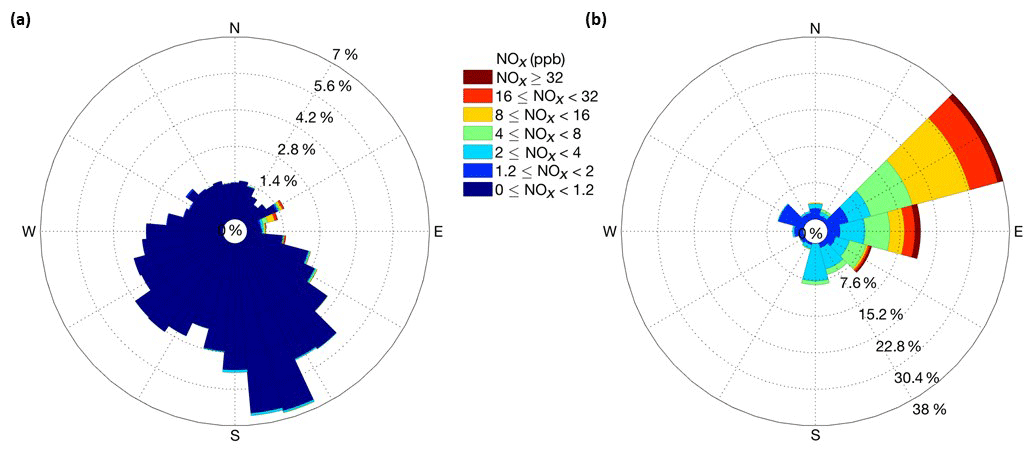
Figure 5(a) Concentration wind rose with the relative frequency of NOx mixing ratio data from the above-surface inlets of the two snow towers segregated by 10∘ sectors for the full year of observation data. This panel shows all data. (b) The same analysis, with wind direction data binned in 20∘ sectors for events when NOx in ambient air exceeded 1.2 ppb.
One of these elevated NOx events is further investigated in Fig. 6. Here, we show the measurements from six inlets on each of the two snow towers over a 1-week period. The sampling of a polluted plume is first observed in the two above-surface inlets (orange/red colored data; +10 and +45 cm), by the sudden increase in NOx from well below 1 ppb to a mixing ratio of ≈13 ppb. This spike in NOx lasted for ≈3 h. After that time, NOx in air sampled above the surface dropped very quickly and equilibrated to prior mixing ratios within less than 0.5 h.
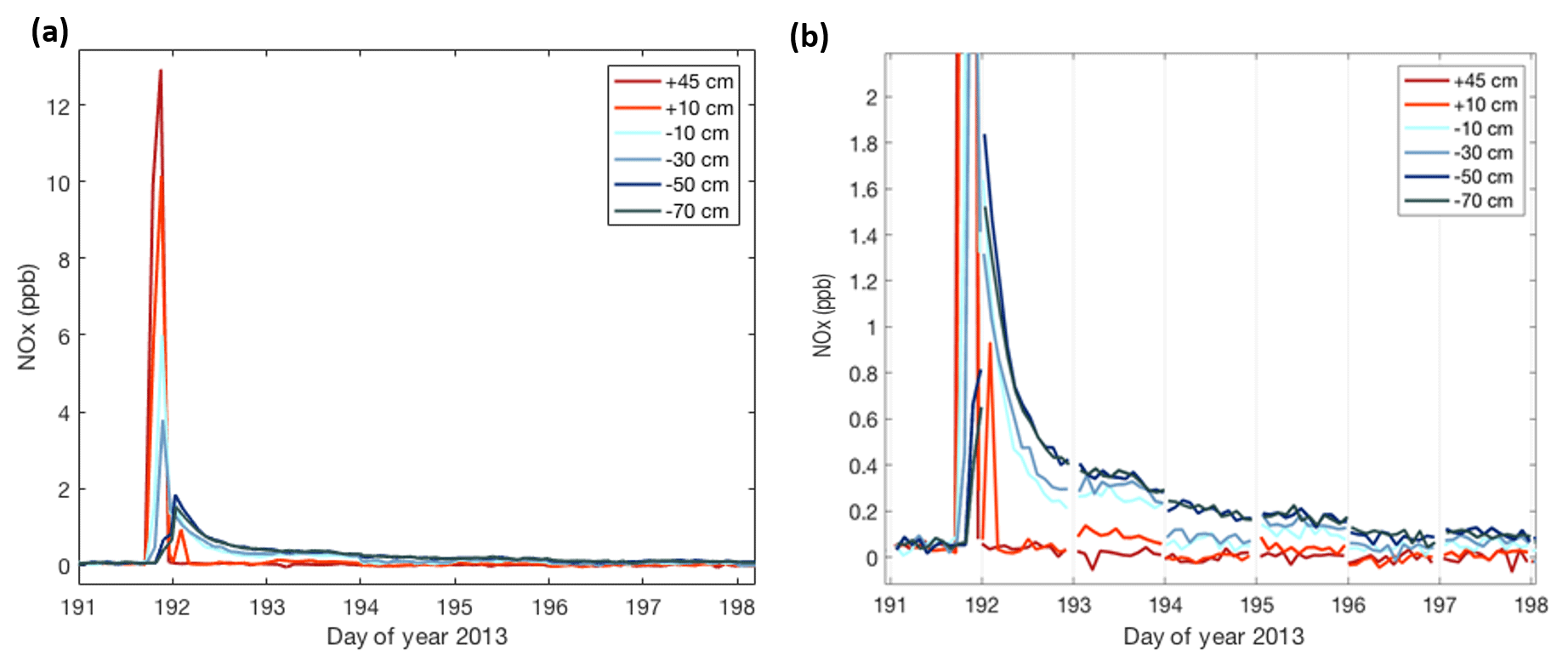
Figure 6Combined measurements from the two snow towers capturing a pollution event at Concordia during the middle of the winter (day of year 191 = 11 July). Plotted time series traces correspond to the sampling heights indicated in the legend, with positive numbers giving the height above the snow surface and negative numbers the depth below the snow surface. Panel (b) is an enlargement of the data shown in panel (a) with the transitions between the connecting lines between two snow tower measurements removed to show the level of agreement in the data from the two sampling manifolds. The sampling switched between the two snow towers every 24 h, leading to some abrupt shifts in the snowpack NOx measurements.
A much different behavior was found in the air sampled from within the snowpack, indicated by the data in the shades of blue. The onset of the pollution signal is delayed, by 1–3 h, with progressively later times deeper in the snowpack. Maximum mixing ratios that are reached in the snowpack are lower, i.e., 10 %–50 % of those that were measured above the surface, with mixing ratio maxima becoming progressively smaller with increasing depth. The most remarkable difference between the above- and below-surface measurements is the longer residence time of the pollution signal in the snowpack. NOx mixing ratios in air withdrawn from all sampling inlets in the snowpack dropped steadily, but remained elevated in comparison to levels seen before the pollution event for ≈1 week. The behavior seen in the measurements from snow tower 1 were in full qualitative agreement and within ≈30 % quantitative agreement with the observations from the second snow tower. After the pollution event, NOx in the snowpack air steadily declined over several days (Fig. 6). Fitting of the data to an exponential decay function yields similar results for all snowpack depths (Fig. 7), with exponential regression fit R2 results of ⩾0.95.
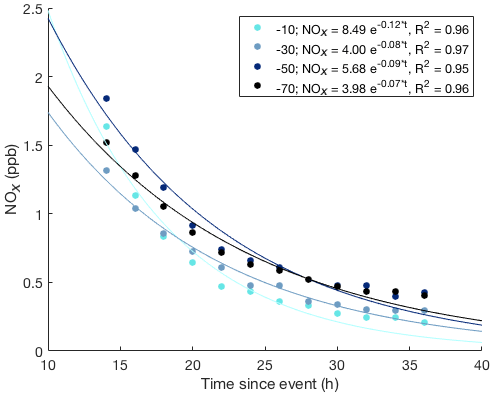
Figure 7Exponential decay function fits to the NOx snowpack measurements versus time at four depths for the event starting on day of year 191 shown in Fig. 6. The start of the event was defined as the time when high NOx was detected above the snowpack. Solutions for the best fit exponential decay functions are given in the legend.
Effects of the exhaust transport were also observed in the ozone signal. The ozone record, shown in Fig. 8, shows a plethora of short positive and negative spikes superimposed on the annual cycle. The up to 15–20 ppb sudden ozone increases seen during the austral summer months are attributable to the photochemical ozone production events that occur in the surface layer of the Antarctic Plateau (see discussion in the introduction section). Occurrences of these elevated ozone events at Concordia Station have previously been investigated by Legrand et al. (2009) and Cristofanelli et al. (2018). In addition to these positive ozone spikes, this annual record also shows numerous sudden negative ozone changes that can be attributed to destruction of ozone by titration of NO in the exhaust plume. Up to 50 % of the ambient ozone was destroyed in air sampled from the above-surface inlets. Similar to NOx, this signal, albeit weaker and attenuated in time, was also seen in the air sampled from within the snowpack (Fig. 8).
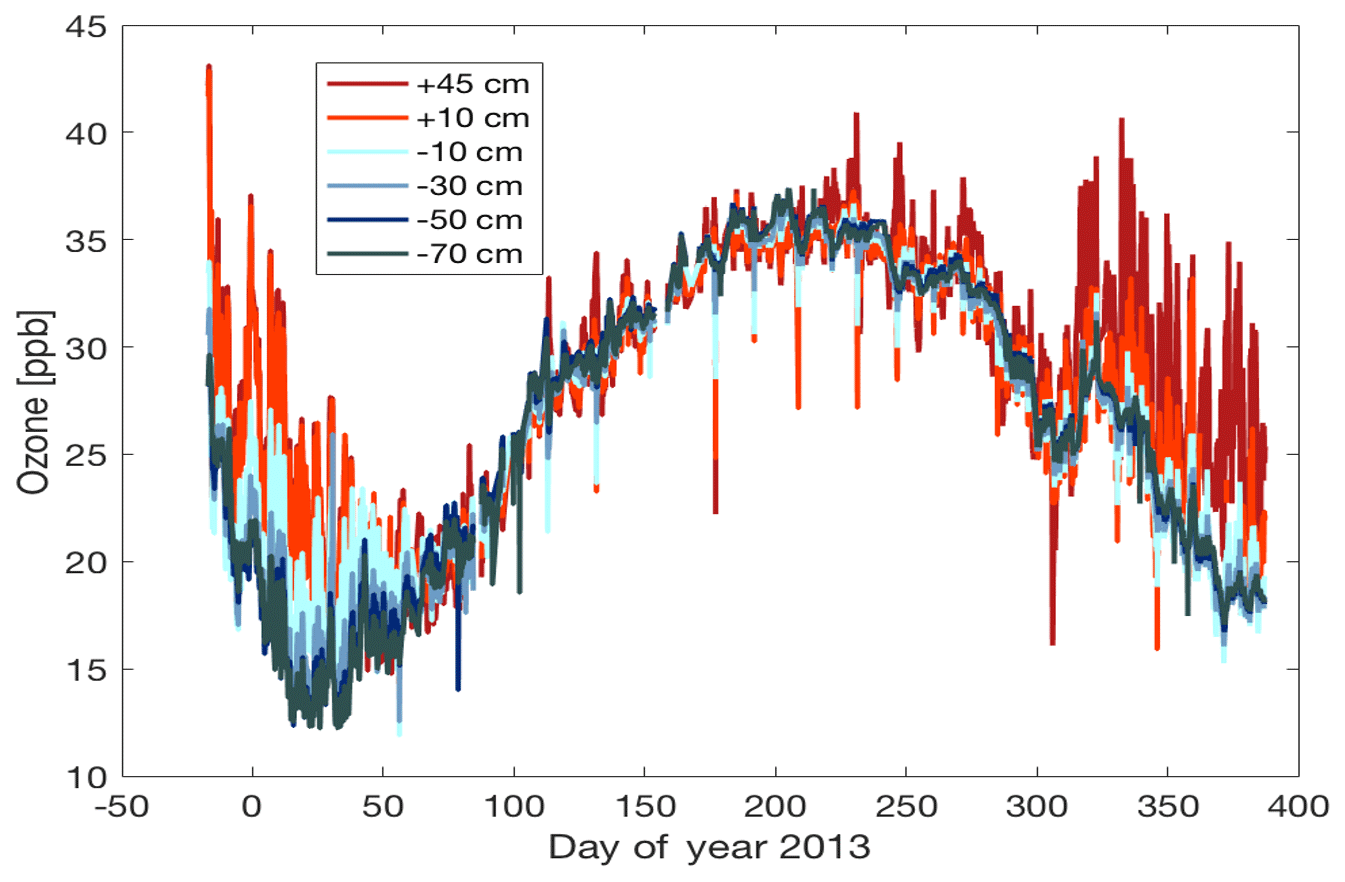
Figure 8Ozone measured from the snow tower inlets throughout the year. Negative spikes in the data coincide with elevated NOx from exhaust infiltration in the snowpack.
Enhancements in formaldehyde in ambient samples suspected to be influenced by station emissions have previously been noted in measurements taken during the OPALE campaign (Preunkert et al., 2015). We therefore investigated if pollution signatures were present in formaldehyde measurements taken in the record from our study. Furthermore, we revisited the GEM measurements taken during the campaign (Angot et al., 2016). Both of these measurements did not quite have the same time resolution and sensitivity as the NOx and ozone monitoring. Formaldehyde measurements overlapped with the ozone and NOx monitoring only for a short period during the 2012/2013 austral summer, and this measurement period suffered from a number of instrument problems. The remaining data did not allow a conclusive evaluation on the behavior of formaldehyde in the snowpack during pollution events. Similarly, we did not identify a clear signature of GEM changes in snowpack air that correlated with the NOx enhancements during pollution events.
Nitrogen oxides undergo reaction with atmospheric oxidants, primarily the OH radical (summer only) and ozone, yielding higher oxidized nitrogen species that can partition into the snowpack aqueous and solid phases. The frequency, large enhancement, and long duration (in the snowpack) of NOx pollution events constitute an apparent unnatural source of NOx to the snowpack. One can hypothesize that reaction of NO and NO2 with OH may be a source of HNO2 and HNO3 in the snowpack, which would add acidity to the snow. This then also poses the question of whether and to what degree photochemical processes, building for instance on or as a substrate, may be altered from natural conditions. However, the overall quantitative effect is likely relatively minor given the large overall reservoir in the solid phase of the snowpack.
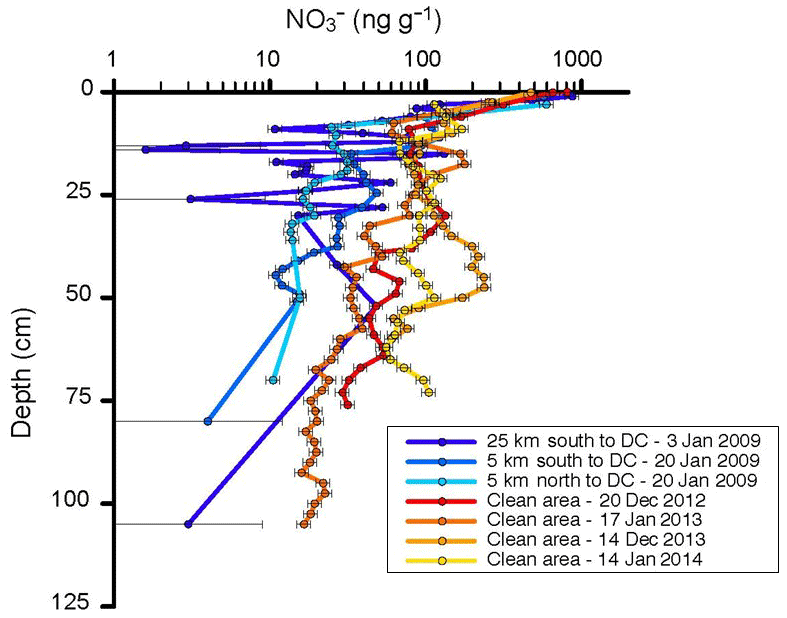
Figure 9Nitrate concentration in snow pits in proximity (≈20 m) to the snow towers at the border of the clean air sector (warm colors), and sampling locations up- and downwind of Concordia (cold colors). Sampling dates are indicated in the figure legend. Horizontal error bars depict the estimated uncertainty of the chemical analysis, i.e., 10 % at >10 ng g−1, 50 % between 5 and 10 ng g−1, and 100 % at <5 ng g−1.
We investigated this question by comparing results from snow pit sampling at different locations within the camp and at up to a 25 km distance from Concordia Station. Nitrate in the snowpack shows a steep vertical gradient, with the highest levels observed right at the surface and progressively lower concentrations with increasing depth (see Fig. 9 and Supplement Fig. S1 for a summary graph where data from both groups were combined and binned in 15 cm depth intervals). The results from the seven snow pits are consistent in the depth profile; however, there appears to be a tendency that the four snow pits within the camp have somewhat higher as well as more variable snow . Deeper into the snowpack, the difference between the two groups of data becomes stronger. At 1 m depth, the snow age is approximately 10 years and therefore approximately corresponds to the start of permanent and year-round activities in 2005 (note that summer activities at Concordia Station were established in 1996). Similar vertical concentration profiles in the Antarctic snowpack have previously been documented (France et al., 2011). Further, relatively higher concentrations (approximately 5 times) are observed in the upper layers of the snowpack during the Antarctic summer (Supplement Fig. S2) than during the winter. This enhancement in surface snow and the seasonal cycle have been linked to the production of HNO3 in the photoactive summer months from reaction of OH with NO2, with the enhancement being the result of HNO3 deposition to the snow surface (Erbland et al., 2013). Elevated concentrations observed at the surface of the snowpack are a common feature in low-accumulation regions, with the concentration values depending strongly on the strategy for collection of the first few centimeters of the snowpack (Erbland et al., 2013; Shi et al., 2018). With the sensitivity of surface snow to the seasonal cycle and the high variability of observed in surface snow and vertical profiles, the currently available data do not allow a conclusive quantitative assessment of the degree to which the snowpack at Concordia is chemically impaired by ventilation of the snowpack with pollution-NOx-enriched air. Contamination of the snowpack around Concordia Station has been noted in previous investigations. Warren et al. (2006) reported an >3-fold increase in snowpack black carbon concentration after the station was established in 2003, with black carbon levels in pre-2003 snow (from deeper depths) also being in closer agreement with snow collected a further distance from the station. Black carbon is one of the contributing factors for a decrease in light penetration into the snowpack (Warren et al., 2006; France et al., 2011; Libois et al., 2013). Consequently, the increased presence of black carbon causes a shallower e-folding depth compared to pristine, uncontaminated snow. In addition to the experiments described above, during the 2014 campaign a number of dynamic flow-through snow chamber photochemistry experiments were conducted to investigate if there were differences in the reactive chemistry in the snow from near the snow tower site compared to snow sampled 25 km away from camp. These measurements showed on the order of 10 %–20 % higher NOx and less ozone in the outflow of chambers filled with the snow from further away from the camp. We did not conduct a high enough number of repeats for evaluating the repeatability and statistical significance of these results to gauge whether and how much of this signal was due to the experimental setup or due to differences in the snowpack chemical composition. Nonetheless, these preliminary findings point towards possible differences in the chemical behavior that potentially are linked to differences in the snow sampling locations and contaminant levels resulting from camp influences that warrant further investigation.
With our snowpack sampling manifold we were able to sample snowpack air to a maximum depth of 70 cm below the surface. Up to ≈2 ppb enhancements in NOx were observed at that depth from exhaust infiltration. While our experiment was not able to access air deeper (than 70 cm), the observed concentration gradients imply that this transport and contamination extend well beyond the depth that was probed in these measurements.
This experiment was a one-spot measurement, at ≈1 km from the camp main facilities. We have no data that would allow us to assess to what distance from the camp the snowpack pollution from exhaust infiltration would be noticeable and of importance, but it likely extends well beyond the distance of our site.
Several other previous studies have noted challenges in sampling clean air at polar research sites that stem from pollution caused by camp exhaust. For atmospheric sampling, this interference can be mitigated by careful postprocessing/filtering of the monitoring data, or by interrupting the sampling during unfavorable wind conditions, which is particularly critical for integrated aerosol collection (Wolff and Cachier, 1998; Wolff et al., 1998). Our experiments from Concordia Station emphasize the pronounced and longer-lasting influence that station exhaust can have on NOx levels inside the snowpack air (compared to ambient air). A tendency of potentially enhanced snowpack levels in two snow pits collected at the camp, compared to data from three sites at a further distance, supports the suspicion that the snowpack chemical oxidized nitrogen composition at the station may be compromised (i.e., contaminated) from the re-occurring ventilation of the snowpack with polluted air. A similar conclusion was derived from δ15N nitrate analyses of snow at Summit: samples collected in the predominant downwind direction of the station generator showed an isotopic signature that had a stronger association with engine exhaust than samples collected at a further distance (Fibiger et al., 2016). Even in the Summit clean air sector, elemental carbon in snow was 1.8–2.4 times higher than in snow collected 10–20 km from the camp (Hagler et al., 2008). The associations shown in our study argue for further investigation, for instance by a high-resolution spatial survey of surface snow composition within and beyond camp boundaries. Given the strong seasonality of , this survey should be performed with as close as possible concurrently conducted snow sampling at selected locations to minimize the influence of temporal changes on the signature.
These observations emphasize concerns about the representativeness of experimental snow chemistry data collected within a polar research camp periphery. This raises the question of how interpretations from such experiments reflect conditions in the remote polar environment. Furthermore, our findings should motivate comparison studies with sampling along transects at a further distance from the main camp facilities. Comparison of these observations will likely yield new insights for evaluating prior polar research site observations and interpretation of snow photochemistry in the glacial snowpack.
Data are available at the Arctic Data Center (https://doi.org/10.18739/A2FP03, Helmig, 2017).
The supplement related to this article is available online at: https://doi.org/10.5194/tc-14-199-2020-supplement.
DH oversaw the study, participated in fieldwork, conducted data analyses and quality control, and prepared the manuscript. DL conducted data analyses and quality control, prepared figures, and contributed to the paper preparation. JH fabricated the instrumental equipment and data acquisition system and participated in the fieldwork. JS contributed to the study design, participated in fieldwork, conducted data analyses and interpretation, and contributed to the manuscript preparation.
The authors declare that they have no conflict of interest.
We thank all staff and scientists who helped with the setup and maintenance of the experiment at Concordia Station. Alexandra Mass, William Vicars, and Albane Barbero helped with some of the field experimental work and data processing. Albane Barbero provided the photograph in Fig. 3.
The French partners recognize technical support from the C2FN (French National Center for Coring and Drilling, handled by INSU), a grant from Labex OSUG@2020 (Investissements d'avenir – ANR10 LABX56), and the program 1011 SUNITEDC of the Institut Polaire Paul Emile Victor (IPEV). The US scientists were supported through a grant from the National Science Foundation (NSF), PLR#1142145.
This paper was edited by Martin Schneebeli and reviewed by two anonymous referees.
Angot, H., Magand, O., Helmig, D., Ricaud, P., Quennehen, B., Gallée, H., Del Guasta, M., Sprovieri, F., Pirrone, N., Savarino, J., and Dommergue, A.: New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale, Atmos. Chem. Phys., 16, 8249–8264, https://doi.org/10.5194/acp-16-8249-2016, 2016.
Crawford, J. H., Davis, D. D., Chen, G., Buhr, M., Oltmans, S., Weller, R., Mauldin, L., Eisele, F., Shetter, R., Lefer, B., Arimoto, R., and Hogan, A.: Evidence for photochemical production of ozone at the South Pole surface, Geophys. Res. Lett., 28, 3641–3644, https://doi.org/10.1029/2001gl013055, 2001.
Cristofanelli, P., Putero, D., Bonasoni, P., Busetto, M., Calzolari, F., Camporeale, G., Grigioni, P., Lupi, A., Petkov, B., Traversi, R., Udisti, R., and Vitale, V.: Analysis of multi-year near-surface ozone observations at the WMO/GAW “Concordia” station (75 degrees 06′ S, 123 degrees 20′ E, 3280 m a.s.l. – Antarctica), Atmos. Environ., 177, 54–63, https://doi.org/10.1016/j.atmosenv.2018.01.007, 2018.
Dassau, T. M., Sumner, A. L., Koeniger, S. L., Shepson, P. B., Yang, J., Honrath, R. E., Cullen, N. J., Steffen, K., Jacobi, H. W., Frey, M., and Bales, R. C.: Investigation of the role of the snowpack on atmospheric formaldehyde chemistry at Summit, Greenland, J. Geophys. Res., 107, 4394, https://doi.org/10.1029/2002JD002182, 2002.
Dibb, J. E., Arsenault, M., Peterson, M. C., and Honrath, R. E.: Fast nitrogen oxide photochemistry in Summit, Greenland snow, Atmos. Environ., 36, 2501–2511, 2002.
Dibb, J. E., Albert, M., Courville, Z., Anastasio, C., Galbavy, E. S., Atlas, E., Beyersdorf, A. J., Blake, D. R., Meinardi, S., Rowland, F. S., Swanson, A. L., Blake, N. J., Bocquet, F., Cohen, L., Helmig, D., Burkhart, J. F., Frey, M. M., Friel, D. K., Hutterli, M. A., Chen, G., Conway, T. J., and Oltrnans, S. J.: An overview of air-snow exchange at Summit, Greenland: Recent experiments and findings, Atmos. Environ., 41, 4995–5006, 2007a.
Dibb, J. E., Whitlow, S. I., and Arsenault, M.: Seasonal variations in the soluble ion content of snow at Summit. Greenland: Constraints from three years of daily surface snow samples, Atmos. Environ., 41, 5007–5019, https://doi.org/10.1016/j.atmosenv.2006.12.010, 2007b.
Eisele, F. L. and Davis, D. D.: Antarctic tropospheric chemistry investigation (ANTCI) 2003, Atmos. Environ., 42, 2747–2748, https://doi.org/10.1016/j.atmosenv.2007.09.074, 2008.
Erbland, J., Vicars, W. C., Savarino, J., Morin, S., Frey, M. M., Frosini, D., Vince, E., and Martins, J. M. F.: Air–snow transfer of nitrate on the East Antarctic Plateau – Part 1: Isotopic evidence for a photolytically driven dynamic equilibrium in summer, Atmos. Chem. Phys., 13, 6403–6419, https://doi.org/10.5194/acp-13-6403-2013, 2013.
Fibiger, D. L., Dibb, J. E., Chen, D., Thomas, J. L., Burkhart, J. F., Huey, L. G., and Hastings, M. G.: Analysis of nitrate in the snow and atmosphere at Summit, Greenland: Chemistry and transport, J. Geophys. Res.-Atmos., 121, 5010–5030, https://doi.org/10.1002/2015jd024187, 2016.
France, J. L., King, M. D., Frey, M. M., Erbland, J., Picard, G., Preunkert, S., MacArthur, A., and Savarino, J.: Snow optical properties at Dome C (Concordia), Antarctica; implications for snow emissions and snow chemistry of reactive nitrogen, Atmos. Chem. Phys., 11, 9787–9801, https://doi.org/10.5194/acp-11-9787-2011, 2011.
Frey, M. M., Savarino, J., Morin, S., Erbland, J., and Martins, J. M. F.: Photolysis imprint in the nitrate stable isotope signal in snow and atmosphere of East Antarctica and implications for reactive nitrogen cycling, Atmos. Chem. Phys., 9, 8681–8696, https://doi.org/10.5194/acp-9-8681-2009, 2009.
Frey, M. M., Brough, N., France, J. L., Erland, J., King, M. D., Savarino, J., Anderson, P., Jones, A., and Wolff, E. W.: Atmospheric nitrogen oxides (NO and NO2) at Dome C: first observations & implications for reactive nitrogen cycling above the East Antarctic Ice Sheet, presented at Air-Ice Chemistry Interactions workshop, 6–7 June, Columbia University, New York, USA, 2011.
Frey, M. M., Brough, N., France, J. L., Anderson, P. S., Traulle, O., King, M. D., Jones, A. E., Wolff, E. W., and Savarino, J.: The diurnal variability of atmospheric nitrogen oxides (NO and NO2) above the Antarctic Plateau driven by atmospheric stability and snow emissions, Atmos. Chem. Phys., 13, 3045–3062, https://doi.org/10.5194/acp-13-3045-2013, 2013.
Frey, M. M., Roscoe, H. K., Kukui, A., Savarino, J., France, J. L., King, M. D., Legrand, M., and Preunkert, S.: Atmospheric nitrogen oxides (NO and NO2) at Dome C, East Antarctica, during the OPALE campaign, Atmos. Chem. Phys., 15, 7859–7875, https://doi.org/10.5194/acp-15-7859-2015, 2015.
Grannas, A. M., Jones, A. E., Dibb, J., Ammann, M., Anastasio, C., Beine, H. J., Bergin, M., Bottenheim, J., Boxe, C. S., Carver, G., Chen, G., Crawford, J. H., Dominé, F., Frey, M. M., Guzmán, M. I., Heard, D. E., Helmig, D., Hoffmann, M. R., Honrath, R. E., Huey, L. G., Hutterli, M., Jacobi, H. W., Klán, P., Lefer, B., McConnell, J., Plane, J., Sander, R., Savarino, J., Shepson, P. B., Simpson, W. R., Sodeau, J. R., von Glasow, R., Weller, R., Wolff, E. W., and Zhu, T.: An overview of snow photochemistry: evidence, mechanisms and impacts, Atmos. Chem. Phys., 7, 4329–4373, https://doi.org/10.5194/acp-7-4329-2007, 2007.
Hagler, G. S. W., Bergin, M. H., Smith, E. A., Town, M., and Dibb, J. E.: Local anthropogenic impact on particulate elemental carbon concentrations at Summit, Greenland, Atmos. Chem. Phys., 8, 2485–2491, https://doi.org/10.5194/acp-8-2485-2008, 2008.
Helmig, D.: Reactive gases in the Dome C snowpack, Antarctica, 2012–2014, Arctic Data Center, https://doi.org/10.18739/A2FP03, 2017.
Helmig, D., Bocquet, F., Cohen, L., and Oltmans, S. J.: Ozone uptake to the polar snowpack at Summit, Greenland, Atmos. Environ., 41, 5061–5076, https://doi.org/10.1016/j.atmosenv.2006.06.064, 2007a.
Helmig, D., Oltmans, S. J., Carlson, D., Lamarque, J. F., Jones, A., Labuschagne, C., Anlauf, K., and Hayden, K.: A review of surface ozone in the polar regions, Atmos. Environ., 41, 5138–5161, https://doi.org/10.1016/j.atmosenv.2006.09.053, 2007b.
Helmig, D., Johnson, B., Oltmans, S. J., Neff, W., Eisele, F., and Davis, D. D.: Elevated ozone in the boundary layer at South Pole, Atmos. Environ., 42, 2788–2803, https://doi.org/10.1016/j.atmosenv.2006.12.032, 2008a.
Helmig, D., Johnson, B. J., Warshawsky, M., Morse, T., Neff, W. D., Eisele, F., and Davis, D. D.: Nitric oxide in the boundary-layer at South Pole during the Antarctic Tropospheric Chemistry Investigation (ANTCI), Atmos. Environ., 42, 2817–2830, 2008b.
Helmig, D., Hueber, J., Liptzin, D., and Savarino, J.: Photo-reactive gases in the snowpack, atmosphere, and their surface exchanges at Dome C, Antarctica, J. Geophys. Res., in preparation, 2020.
Honrath, R. E., Peterson, M. C., Guo, S., Dibb, J. E., Shepson, P. B., and Campbell, B.: Evidence of NOx production within or upon ice particles in the Greenland snowpack, Geophys. Res. Lett., 26, 695–698, 1999.
Honrath, R. E., Lu, Y., Peterson, M. C., Dibb, J. E., Arsenault, M. A., Cullen, N. J., and Steffen, K.: Vertical fluxes of NOx, HONO, and HNO3 above the snowpack at Summit, Greenland, Atmos. Environ., 36, 2629–2640, 2002.
Jacobi, H. W., Bales, R. C., Honrath, R. E., Peterson, M. C., Dibb, J. E., Swanson, A. L., and Albert, M. R.: Reactive trace gases measured in the interstitial air of surface snow at Summit, Greenland, Atmos. Environ., 38, 1687–1697, https://doi.org/10.1016/j.atmosenv.2004.01.004, 2004.
Jones, A. E., Weller, R., Wolff, E. W., and Jacobi, H. W.: Speciation and rate of photochemical NO and NO2 production in Antarctic snow, Geophys. Res. Lett., 27, 345–348, 2000.
Jones, A. E., Weller, R., Anderson, P. S., Jacobi, H. W., Wolff, E. W., Schrems, O., and Miller, H.: Measurements of NOx emissions from the Antarctic snowpack, Geophys. Res. Lett., 28, 1499–1502, 2001.
Jones, A. E., Wolff, E. W., Salmon, R. A., Bauguitte, S. J.-B., Roscoe, H. K., Anderson, P. S., Ames, D., Clemitshaw, K. C., Fleming, Z. L., Bloss, W. J., Heard, D. E., Lee, J. D., Read, K. A., Hamer, P., Shallcross, D. E., Jackson, A. V., Walker, S. L., Lewis, A. C., Mills, G. P., Plane, J. M. C., Saiz-Lopez, A., Sturges, W. T., and Worton, D. R.: Chemistry of the Antarctic Boundary Layer and the Interface with Snow: an overview of the CHABLIS campaign, Atmos. Chem. Phys., 8, 3789–3803, https://doi.org/10.5194/acp-8-3789-2008, 2008.
Kramer, L. J., Helmig, D., Burkhart, J. F., Stohl, A., Oltmans, S., and Honrath, R. E.: Seasonal variability of atmospheric nitrogen oxides and non-methane hydrocarbons at the GEOSummit station, Greenland, Atmos. Chem. Phys., 15, 6827–6849, https://doi.org/10.5194/acp-15-6827-2015, 2015.
Legrand, M., Preunkert, S., Jourdain, B., Gallee, H., Goutail, F., Weller, R., and Savarino, J.: Year-round record of surface ozone at coastal (Dumont d'Urville) and inland (Concordia) sites in East Antarctica, J. Geophys. Res., 114, 1–12, https://doi.org/10.1029/2008jd011667, 2009.
Legrand, M., Preunkert, S., Savarino, J., Frey, M. M., Kukui, A., Helmig, D., Jourdain, B., Jones, A. E., Weller, R., Brough, N., and Gallée, H.: Inter-annual variability of surface ozone at coastal (Dumont d'Urville, 2004–2014) and inland (Concordia, 2007–2014) sites in East Antarctica, Atmos. Chem. Phys., 16, 8053–8069, https://doi.org/10.5194/acp-16-8053-2016, 2016.
Libois, Q., Picard, G., France, J. L., Arnaud, L., Dumont, M., Carmagnola, C. M., and King, M. D.: Influence of grain shape on light penetration in snow, The Cryosphere, 7, 1803–1818, https://doi.org/10.5194/tc-7-1803-2013, 2013.
Molina, M. J. and Molina, L. T.: Megacities and atmospheric pollution, J. Air Waste Manage., 54, 644–680, 2004.
Murray, K. A., Kramer, L. J., Doskey, P. V., Ganzeveld, L., Seok, B., Van Dam, B., and Helmig, D.: Dynamics of ozone and nitrogen oxides at Summit, Greenland. II. Simulating snowpack chemistry during a spring high ozone event with a 1-D process-scale model, Atmos. Environ., 117, 110–123, https://doi.org/10.1016/j.atmosenv.2015.07.004, 2015.
Neff, W., Helmig, D., Grachev, A., and Davis, D.: A study of boundary layer behavior associated with high NO concentrations at the South Pole using a minisodar, tethered balloon, and sonic anemometer, Atmos. Environ., 42, 2762–2779, 2008.
Preunkert, S., Ancellet, G., Legrand, M., Kukui, A., Kerbrat, M., Sarda-Esteve, R., Gros, V., and Jourdain, B.: Oxidant Production over Antarctic Land and its Export (OPALE) project: An overview of the 2010–2011 summer campaign, J. Geophys. Res., 117, 6689–6705, https://doi.org/10.1029/2011jd017145, 2012.
Preunkert, S., Legrand, M., Frey, M. M., Kukui, A., Savarino, J., Gallée, H., King, M., Jourdain, B., Vicars, W., and Helmig, D.: Formaldehyde (HCHO) in air, snow, and interstitial air at Concordia (East Antarctic Plateau) in summer, Atmos. Chem. Phys., 15, 6689–6705, https://doi.org/10.5194/acp-15-6689-2015, 2015.
Seok, B., Helmig, D., Williams, M. W., Liptzin, D., Chowanski, K., and Hueber, J.: An automated system for continuous measurements of trace gas fluxes through snow: an evaluation of the gas diffusion method at a subalpine forest site, Niwot Ridge, Colorado, Biogeochemistry, 95, 95–113, https://doi.org/10.1007/s10533-009-9302-3, 2009.
Shi, G., Hastings, M. G., Yu, J., Ma, T., Hu, Z., An, C., Li, C., Ma, H., Jiang, S., and Li, Y.: Nitrate deposition and preservation in the snowpack along a traverse from coast to the ice sheet summit (Dome A) in East Antarctica, The Cryosphere, 12, 1177–1194, https://doi.org/10.5194/tc-12-1177-2018, 2018.
Steinbacher, M., Zellweger, C., Schwarzenbach, B., Bugmann, S., Buchmann, B., Ordonez, C., Prevot, A. S. H., and Hueglin, C.: Nitrogen oxide measurements at rural sites in Switzerland: Bias of conventional measurement techniques, J. Geophys. Res., 112, D11307, https://doi.org/10.1029/2006jd007971, 2007.
Van Dam, B., Helmig, D., Toro, C., Doskey, P., Kramer, L., Murray, K., Ganzeveld, L., and Seok, B.: Dynamics of ozone and nitrogen oxides at Summit, Greenland: I. Multi-year observations in the snowpack, Atmos. Environ., 123, 268–284, https://doi.org/10.1016/j.atmosenv.2015.09.060, 2015.
Warren, S. G., Brandt, R. E., and Grenfell, T. C.: Visible and near-ultraviolet absorption spectrum of ice from transmission of solar radiation into snow, Appl. Optics, 45, 5320–5334, https://doi.org/10.1364/ao.45.005320, 2006.
Wolff, E. W. and Cachier, H.: Concentrations and seasonal cycle of black carbon in aerosol at a coastal Antarctic station, J. Geophys. Res.-Atmos., 103, 11033–11041, https://doi.org/10.1029/97jd01363, 1998.
Wolff, E. W., Legrand, M. R., and Wagenbach, D.: Coastal Antarctic aerosol and snowfall chemistry, J. Geophys. Res.-Atmos., 103, 10927–10934, https://doi.org/10.1029/97jd03454, 1998.